Cancer is a complex cellular mechanism that occurs at least by a mutation of five or six genes, each mutation alone causes changes in the cell.1,2 Colorectal cancer (CRC) is one of the most common gastrointestinal cancers and has high mortality rates.3 The formation of tumors in the rectum, colon, and appendix and the extensive and advanced accumulation of genetic and epigenetic changes alter the natural epithelium of the colon to adenoma and ultimately become a malignant tumor.4 As a result of genetic and epigenetic changes, colon mucosal cells change from normal to cancerous cells.5 Cancer disrupts the cellular order, and this cellular disturbance directly affects the cell cycle and causes a lack of cell differentiation. An increase in the number of cancer patients and in the average age of the population directly correlates with the increase in cancer in the world. In this regard, the genetic and epigenetic study of different molecular pathways involved in CRC can be beneficial for early diagnosis and treatment.6–10 Many genes are involved in different molecular mechanisms in the carcinogenic pathway, including MYC, SMAD2/3, and DNMT3A. In this review, we discuss the performance of MYC, SMAD2/3, and DNMT3A and the role they play in carcinogenesis.11,12
Fluctuation of genes
MYC is transmitted by the avian myelocytomatosis virus, and viral promoters widely regulate the extent of the gene.13 Some noticeable changes occur in the expression of MYC oncogene and increase the process of cell deformation. At least three different mechanisms can produce MYC oncogene. In several human tumors, the amount of MYC gene is determined by its natural expression promoters. Still, the number of copies of this gene is several times more than the number of copies in the normal human genome.14 In 30% of children’s neuroblastoma, a similar gene called N-MYC is also widely distributed in malignant tumors [Figure 1]. In both cases, the increase in these genetic copies increases the level of the produced gene. Another point is that MYC family proteins have a very significant effect on cell growth.15 Consequently, when they are present in large quantities, they cause uncontrolled cell proliferation. MYC proto-oncogenes, commonly referred to as C-MYC, are distinct from the two N-MYC and L-MYC genes. Human MYC genes are seen in a variety of human tumors.16 In addition to genetic and environmental factors, epigenetic factors play a vital role in carcinogenesis. These factors include histone changes, acetylation, phosphorylation, and DNA methylation.17,18
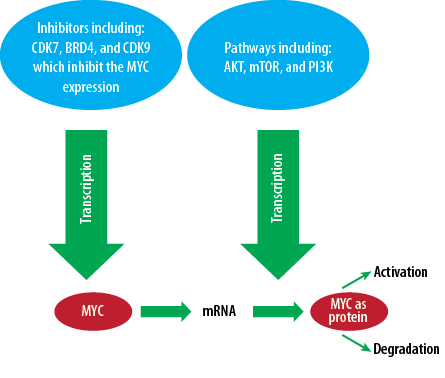
Figure 1: Significance of inhibitors like CDK7, BRD4, and CDK9 in pathways including AKT, mTOR, and PI3K, which inhibit MYC expression.
DNA methylation, an essential epigenetic agent, is a common feature in vertebrates, and one of the main epigenetic mechanisms is the control of gene expression.19 Methylation changes can be eliminated or transferred to the next generation without changing the nature of the DNA. Also, CpG methylation is one of the most critical molecular processes in carcinogenesis. The study of promoter hypermethylation can create new hopes and achievements to achieve molecular diagnostic markers of cancer.
In the case of the SMAD gene, the proteins encoded by these genes belonging to the SMAD group of proteins are similar to those of Drosophila melanogaster genes (Elegans SMAD gene).20 The SMAD proteins are a signal transducer and transcription modulator that interfaces multiple signaling pathways. These proteins are the main signal transducers for the transforming growth factor-beta (TGF-β) superfamily receptors, which are critical for regulating cell proliferation, apoptosis, and differentiation.21 In response to the signal, TGF-β superfamily ligands bind to a type II receptor, which recruits and phosphorylates a type I receptor. The type I receptor then phosphorylates receptor-regulated SMADs (R-SMADs), which can now bind coSMAD and SMAD4. R-SMAD/coSMAD complexes accumulate in the nucleus, where they act as transcription factors and participate in regulating target gene expression.22 Besides SMAD2, which is a protein-encoding gene, major diseases associated with SMAD2 include urogenital disease and many common cancers.23 The pivotal paralog of the SMAD2 is the SMAD3 [Figure 2]. Human immunohistochemistry assessment of SMAD3/SMAD2 phosphorylation and p300 activator showed association with human glomerulonephritis and renal injury.24 Also, SMAD2 and SMAD4 mutations in the TGF-β-SMAD signaling pathway have been proven in head and neck carcinoma.25 In addition, the SMAD pathway is also active in scleroderma fibroblast, and the level of SMAD2/3 phosphoryl and the site of the SMAD2/3 phosphoryl was increased.26 SMAD2/3/4 heterodimers correspondingly regulate SMDA2/3/4 transcriptional activity. The SMAD3 gene produces a protein involved in transmitting chemical signals from the cell surface to the nucleus.27 This signaling process begins when TGF-β protein binding to a receptor on the cell surface and activates a group of SMAD proteins such as SMAD2 and SMAD3.28 These SMAD proteins form a complex with SMAD4 and then accumulate in the nucleus and binds to specific regions of DNA to control the activity of particular genes. Through the TGF-β signaling pathway, the SMAD2/3 proteins also affect many aspects of cellular processes, including growth and division (proliferation), cell movement (migration), and cell death (apoptosis).29
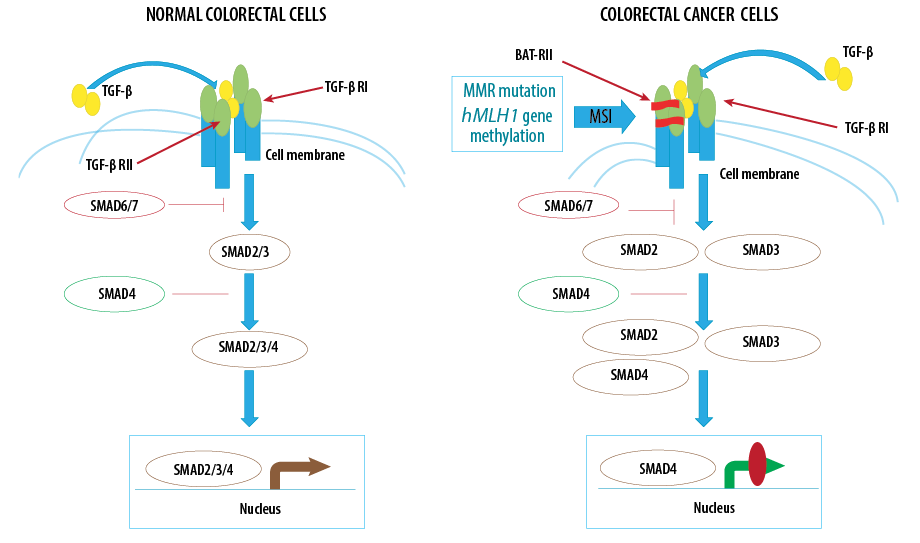
Figure 2: Fluctuations of SMADs (SMAD2/3/4/6 and 7) alongside other related genes in normal and colon cancer cells.
Another enzyme is the DNA (cytosine-5)-methyltransferase 3A (DNMT3A) that is encoded in humans by the DNMT3A gene [Figure 3].30 DNMT3A catalyzes the transfer of methyl groups to specific CpG structures in DNA, a process called DNA methylation.31 DNA methylation plays a role in many cellular functions, such as gene expression regulation, protein and lipid reaction regulation, and chemical processing control in nervous system signaling that by DNMT3A occurs through methylation during evolution.32 This enzyme can also lead to the formation of more mature cell types in the early cells. In early blood stem cells called hematopoietic stem cells, methylation patterns are generated by the DNMT3A, which develops the differentiation to different types of blood cells.33–36
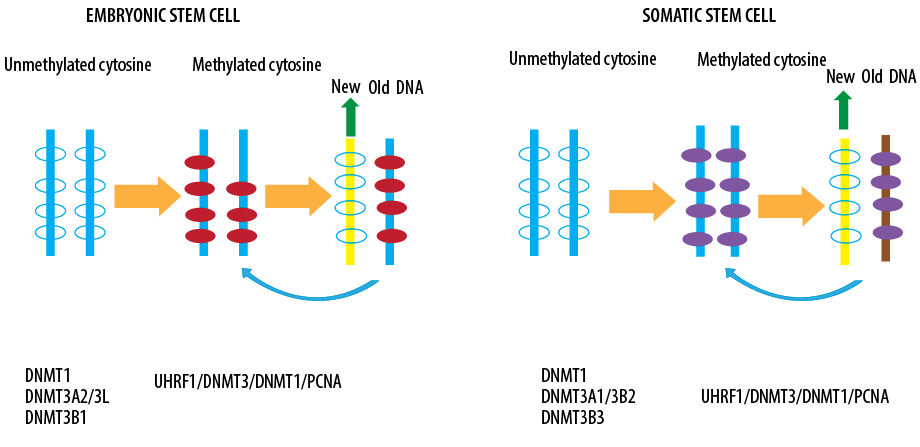
Figure 3: Role of DNA methyltransferases in embryonic and somatic stem cells.
Induction of epigenetics elements in carcinogenesis and tumorogenesis
Epigenetic elements include DNA methylation, histone modification, histone acetylation, histone phosphorylation, and RNA remodeling. The most important is DNA methylation.37 Additionally, all these elements indicated their remarkable and impressive role in carcinogenesis and tumorogenesis.38 Unlike changes related to the main DNA sequence, such as mutations, most epigenetic changes are reversible. Naturally, phenomena such as genomic imprinting, inactivation of the X chromosome, and the expression of gene sets that are important in the process of embryonic development are controlled by epigenetic mechanisms.39,40 In recent decades, studies have shown that epigenetic alteration patterns and genetic changes in some genes play an important role in tumorigenesis. These changes include abnormal methylation patterns in gene regulation, histone modification, and alterations in miRNA expression.41 Recent studies suggest that the abnormal methylation pattern on CpG islands is effective in tumor cell proliferation. The increase in the methylation of the regulatory regions of tumor suppressor genes and DNA repair leads to the extinction of these genes and the development of cancer. On the other hand, the reduction of methylation in the regulatory regions of oncogenes increases their expression and leads to the conduction of cells to tumors.42 This mechanism is involved in the development of cancer cells by activating the enzymes involved in cell growth and survival of apoptosis and the cell cycle.43
Because DNA has an important role in replication and transcription and ultimately cell proliferation, the most important targets are regulatory molecules and anticancer drugs.44
DNA methylation is regulated by the DNA methyltransferase enzyme. Increased expression of DNA methyltransferase appears to be a common feature in a variety of cancers. Methylation patterns are inherited through mitosis. These normal patterns are disrupted in the DNA of the cancer cell; CpG islands are prone to methyltransferase activity and other areas of DNA are hypotensive.45 The hypermethylation profile of CpG islands varies in different genes for each type of cancer. In general, hypermethylation of CpG islands occurs in tumor suppressor genes, and genes involved in the cell cycle, DNA repair, carcinogenic metabolism, intercellular interactions, cell death and regression, and promotes cancer progression.46
Performance of DNA methylation in CRC
Investigating gene expression in gastrointestinal cancers to evaluate their fluctuations, alongside any epigenetics alterations, is of great importance
[Table 1].47–51 Remarkably, the mechanisms underlying CRC pathobiology remain subjects of wide study in the pathogenesis of cancer. Genetic and epigenetic modifications have resulted in CRC and also, the cellular genome that transforms normal glandular epithelium into adenocarcinoma is involved in this process.52,53 Conspicuously, the evaluation of methylated genes in CRC has also revealed a unique molecular subgroup of CRCs called CpG island methylator phenotype cancers; these tumors have a high frequency of methylated genes. In addition to DNA hypermethylation that often takes place in the promoter region of tumor suppressor genes, epigenetic regulation of CRC epigenome also includes histone post-translational modifications, primarily histone acetylation and methylation that also play critical roles in the regulation of expression of oncogenes and tumor suppressor genes.52,54,55 In this way, epigenetics alludes to heritable gene expression modifications that are not mediated by alterations in the DNA sequence. The epigenetic regulation of gene expression happens in normal tissue and plays a key role in many cellular activities, including tissue differentiation, embryonic development, and imprinting.56
Table 1: Main models of involved genomic uncertainty in colon cancer.
|
Sporadic colorectal cancer with mismatch repair deficiency |
MLH1 somatic methylation |
Colorectal cancer with increased risk of poor differentiation, more commonly located in the right colon, less aggressive clinical behavior than tumors without mismatch repair deficiency |
Somatic |
|
Base excision repair defect MYH-associated polyposis |
MYH |
Development of 15 or more colorectal adenomas with increased risk of colorectal cancer |
Germline |
|
Chromosomal instability- loss of heterozygosity at multiple loci |
Loss of heterozygosity at APC, TP53, and SMAD4 |
Characteristic of 80% to 85% of sporadic colorectal cancers, depending on stage |
Somatic |
|
CpG island methylator phenotype-methylation target loci |
Target loci MLH1, MINT1 MINT2, and MINT3 |
Characteristic of 15% of colorectal cancers, with most showing mismatch repair deficiency from loss of tumor MLH1 expression |
Somatic |
In 1982, aberrant epigenetic modifications were first explored in CRC. Noticeably, the epigenetic study has indicated an epigenetic landscape comprising an elaborate array of epigenetic regulatory mechanisms that control gene expression in tumor and non-tumor tissues.57,58 The epigenetic landscape is largely a reflection of agents that ascertain the condensation state of the chromatin, which identifies whether the DNA is related to proteins that manage the gene transcription. A relaxed or open chromatin state permits gene transcription, whereas a condensed chromatin state prevents gene transcription.56 Evidently, it is confirmed that the DNA methylation seen in cancer and aging may stem from a small population of cells. Notably, not only are the target sites found partially methylated in normal tissues but are also highly altered in polyps,59 a very early stage in the generation of colon cancer in men. It reveals that a subpopulation of stem cells in the colon undergoes de novo methylation of target CpG islands during aging, which presumably generates small patches of tissue that carry an aberrant DNA methylation profile.60–62 This alteration probably induces a state of constitutive heterochromatin, which is not easily reversible. Thus, proliferative cells in the crypt that transform this sign may have the ability to skip the polycomb structure itself, but would not be capable of activating the critical differentiation genes, thereby inhibiting these cells from undergoing a transition to the epithelium, thus leaving them in a relatively proliferative state. Although this might not be sufficient for generating a tumor, it could very well provide the necessary background for cells that have transformed either through prior genetic predisposition or spontaneous mutation. These particular cells collect and organize DNA methylation during aging and then perform as preferred targets for the transformation process, which is protected by the observation that both polyps and normal tissue surrounding the tumor are highly methylated and by the experimental evidence showing that 5-azacytidine is only capable of preventing the accumulation of intestinal tumors in mice if it originates from early in life.59,63,64 As DNA methylation plays a significant role in CRC formation, it can be used as a potential diagnostic biomarker in cancer detection.65 Considerably, some gene promoter methylation in the plasma or serum of patients with CRC has shown great promise as a potential diagnostic indicator of CRC.
To date, a lot of hypermethylated genes have been reported in CRC, but only a few have been included in commercial blood-based tests. Studies are needed to find new practical biomarkers for prognosis that would aid researchers and practitioners in decision-making. High-throughput technologies, such as methylation microarrays and next generation sequencing, have helped advance our understanding of epigenetic events at the genomic level.66–68
Role of DNA methylation of SMAD2/3, MYC, and DNMT3A in CRC
Aberrant de novo methylation of DNA is considered a remarkable mediator of tumorigenesis. The processes that mediate aberrant DNA hyper- and hypo-methylation are under study. Although certain mechanisms have yet to be identified, it is now clear that DNA methylation is regulated through reciprocal interactions with histones. Modifications in the post-translational state of histones are closely related to cancer-related alterations in DNA methylation.69 The enzymes that mediate DNA methylation, DNMT1, DNMT3A, and DNMT3B, are overexpressed, hyperactive, or misdirected. Increased DNMT expression has been proposed as a mechanism for the increased methylation seen in the promoter region of tumors. Compared to normal tissues, the increased expression and increased function of the DNA methyltransferases (DNMTs) have been reported in human cancers, including colon cancer.70 DNMTs catalyze the addition of a methyl group to the 5-cytosine residue of CpG dinucleotides. This family of enzymes comprises DNMT1 that performs as a DNA maintenance methyltransferase, and DNMT3A and DNMT3B that methylate previously unmethylated regions of DNA and are required for genome-wide de novo DNA methylation. DNMT3A protein expression in human tissue samples and its potential inhibition by DNMT inhibitors remains unclear.71 MYC protein has been implicated in development through the cell cycle and in differentiation-related regulation of transcription. Additionally, overexpressed MYC protein in dysplastic and tumor cells, accumulating in the cytoplasm and transferring persistently to the nucleus, may modify the cellular response to growth factors and abrogate normal growth control mechanisms by controlling cells from escaping from the proliferation cycle.72 Conspicuously, C-MYC protein may manage its expression fluctuation via binding process, directly or indirectly, to the C-MYC gene. If this interaction is influenced by DNA methylation,73,74 there may be a feedback effect between hypomethylation of the third exon of MYC and deregulation of expression. One study indicated that a 34-base pair sequence spanning the CCGG site of the C-MYC third exon exhibits methylation-dependent binding of particular protein species from normal colonic epithelium; dysplastic tissue yields a modified binding pattern.75 Changes in the downstream methylation model may influence MYC expression by binding trans-acting agents, either directly or via induction of long range conformational alterations. The TGF-β pathway plays a key role in embryonic development, organ homeostasis, tissue repair, and disease.76,77 This diversity of tasks is achieved through the intracellular effector SMAD2/3, whose canonical function is to control the activity of target genes by interacting with transcriptional regulators.78 Despite that, a complete description of the factors interacting with SMAD2/3 in any given cell type is still lacking. SMAD2/3 could act as a hub coordinating several proteins known to have a role in mRNA processing and alteration, apoptosis, DNA repair, and transcriptional regulation.79 Remarkably, in the therapeutic approach, a probiotic strategy like using gut microbiota is of great importance.80
The key role of microRNAs in carcinogenesis
Based on the chemical structure of RNA, which is made up of only four flat bases and the nucleotides have a negative charge, it seems that the drug target is not promising. However, RNA molecules can bind to small molecules. The binding of small ligands to RNA by blocking macromolecule binding changes the active RNA configuration, induces a sub-configuration on the RNA, and inhibits the RNA catalytic activity, affecting its biological activity. Some herbal anticancer compounds, such as curcumin in turmeric, interact with RNA. More than 80% of the genome is actively grouped with RNA transcripts, referred to as non-coding RNAs. This group includes tRNA, rRNA, small nuclear RNA involved in splicing, and microRNA.81
MicroRNAs are a type of non-coding RNA that is fully protected nucleotides during evolution. These molecules are induced by binding to 3’UTR inhibiting translation or induction. Epigenetic factors reduce the expression of microRNAs by over-methylation of gene promoters or histone modifications. Increased expression of microRNAs in cancer cells could be due to the proliferation and lack of control of a transcription factor or demethylation of CpG islands in gene promoter areas.82 It is not yet clear whether the change in microRNA expression is the result of a pathological state of cancer or whether cancer is the main cause of these changes. However, many microRNAs, especially the two groups of oncogenic microRNAs and tumor inhibitors, are abnormally expressed in cancer cells.83 Epigenetic factors reduce the expression of microRNAs by overmethylation of gene promoters or histone modifications.
Conclusion
Cancer is a genetic disease that occurs due to sequential mutations in human genes and genetic and environmental factors. CRC is one of the most prevalent lethal forms of cancer worldwide. Several mechanisms are involved in cancer development, which play major roles in altering cellular signaling and cancer formation. Oncogenes, tumor suppressor genes, apoptosis genes, and restorative genes are among the major factors in cancerous cells. These genes are responsible for controlling the differentiation and growth of the cells. Consequently, the mutation in these genes causes the normal process of the cell to become cancerous. Oncogenes are also activated by mutation in the original genes and converted to proto-oncogenes. Mutations in tumor suppressor genes also cause abnormal cell division and transform healthy cells into cancerous ones. Another factor is programmed cell death (apoptosis), which is the last cellular escape from the cancerous process. Therefore, studying all factors involved in carcinogenesis, particularly the changes in epigenetic factors, such as DNA methylation, is useful in identifying diagnostic and therapeutic biomarkers in CRC.
Disclosure
The authors declared no conflicts of interest. No funding was received for this study.
Acknowledgements
The authors express their appreciation and gratitude to all people who were involved in this review.
references
- 1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015 Mar;136(5):E359-E386.
- 2. Samadani AA, Akhavan-Niaki H. Interaction of sonic hedgehog (SHH) pathway with cancer stem cell genes in gastric cancer. Med Oncol 2015 Mar;32(3):48.
- 3. Lin S-H, Raju GS, Huff C, Ye Y, Gu J, Chen J-S, et al. The somatic mutation landscape of premalignant colorectal adenoma. Gut 2018 Jul;67(7):1299-1305.
- 4. Terdiman JP, Aust DE, Chang CG, Willenbucher RF, Baretton GB, Waldman FM. High resolution analysis of chromosome 18 alterations in ulcerative colitis-related colorectal cancer. Cancer Genet Cytogenet 2002 Jul;136(2):129-137.
- 5. Enderle-Ammour K, Bader M, Ahrens TD, Franke K, Timme S, Csanadi A, et al. Form follows function: Morphological and immunohistological insights into epithelial-mesenchymal transition characteristics of tumor buds. Tumour Biol 2017 May;39(5):1010428317705501.
- 6. Norollahi SE, Hamidian SM, Kohpar ZK, Azadi R, Rostami P, Vahidi S, et al. The fluctuation of APC gene in WNT signaling with adenine deletion of adenomatous polyposis coli, is associated in colorectal cancer. J Coloproctol (Rio J) 2020;40(2):135-142.
- 7. Babaei K, Shams S, Keymoradzadeh A, Vahidi S, Hamami P, Khaksar R, et al. An insight of microRNAs performance in carcinogenesis and tumorigenesis; an overview of cancer therapy. Life Sci 2020 Jan;240:117077.
- 8. Akhavan-Niaki H, Samadani AA. Molecular insight in gastric cancer induction: an overview of cancer stemness genes. Cell Biochem Biophys 2014 Apr;68(3):463-473.
- 9. Fattahi S, Pilehchian Langroudi M, Samadani AA, Nikbakhsh N, Asouri M, Akhavan-Niaki H. Application of unique sequence index (USI) barcode to gene expression profiling in gastric adenocarcinoma. J Cell Commun Signal 2017 Mar;11(1):97-104.
- 10. Kosari-Monfared M, Nikbakhsh N, Fattahi S, Ghadami E, Ranaei M, Taheri H, et al. CTNNBIP1 downregulation is associated with tumor grade and viral infections in gastric adenocarcinoma. J Cell Physiol 2019 Mar;234(3):2895-2904.
- 11. Yang X, Zhu F, Yu C, Lu J, Zhang L, Lv Y, et al. N-myc downstream-regulated gene 1 promotes oxaliplatin-triggered apoptosis in colorectal cancer cells via enhancing the ubiquitination of Bcl-2. Oncotarget 2017 Jul;8(29):47709-47724.
- 12. Wang W, Deng J, Wang Q, Yao Q, Chen W, Tan Y, et al. Synergistic role of Cul1 and c-Myc: prognostic and predictive biomarkers in colorectal cancer. Oncol Rep 2017 Jul;38(1):245-252.
- 13. Yang T-W, Gao Y-H, Ma S-Y, Wu Q, Li Z-F. Low-grade slightly elevated and polypoid colorectal adenomas display differential β-catenin-TCF/LEF activity, c-Myc, and cyclin D1 expression. World J Gastroenterol 2017 May;23(17):3066-3076.
- 14. Parang B, Kaz AM, Barrett CW, Short SP, Ning W, Keating CE, et al. BVES regulates c-Myc stability via PP2A and suppresses colitis-induced tumourigenesis. Gut 2017 May;66(5):852-862.
- 15. Mastropasqua F, Marzano F, Valletti A, Aiello I, Di Tullio G, Morgano A, et al. TRIM8 restores p53 tumour suppressor function by blunting N-MYC activity in chemo-resistant tumours. Mol Cancer 2017 Mar;16(1):67.
- 16. Xie F, Yuan Y, Xie L, Ran P, Xiang X, Huang Q, et al. miRNA-320a inhibits tumor proliferation and invasion by targeting c-Myc in human hepatocellular carcinoma. Onco Targets Ther 2017 Feb;10:885-894.
- 17. Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, Uno N, et al. Dynamics of epigenetic regulation at the single-cell level. Science 2016 Feb;351(6274):720-724.
- 18. Tiffon C. The impact of nutrition and environmental epigenetics on human health and disease. Int J Mol Sci 2018 Nov;19(11):3425.
- 19. Balch C, Ramapuram JB, Tiwari AK. The epigenomics of embryonic pathway signaling in colorectal cancer. Front Pharmacol 2017 May;8:267.
- 20. Wotton D, Lo RS, Lee S, Massagué J. A Smad transcriptional corepressor. Cell 1999 Apr;97(1):29-39.
- 21. Lin H, Yang Q, Wang H, Qi J, Zhang H, Wang H, et al. Involvement of SMAD4, but not of SMAD2, in transforming growth factor-beta1-induced trophoblast expression of matrix metalloproteinase-2. Frontiers in Bioscience: A Journal and Virtual Library 2006;11:637-646.
- 22. Wrana JL, Attisano L. The Smad pathway. Cytokine Growth Factor Rev 2000 Mar-Jun;11(1-2):5-13.
- 23. Xu J, Attisano L. Mutations in the tumor suppressors Smad2 and Smad4 inactivate transforming growth factor β signaling by targeting Smads to the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A 2000 Apr;97(9):4820-4825.
- 24. Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, et al. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J 1997 Sep;16(17):5353-5362.
- 25. Ottesen JJ, Huse M, Sekedat MD, Muir TW. Semisynthesis of phosphovariants of Smad2 reveals a substrate preference of the activated T β RI kinase. Biochemistry 2004 May;43(19):5698-5706.
- 26. Cao Z, Flanders KC, Bertolette D, Lyakh LA, Wurthner JU, Parks WT, et al. Levels of phospho-Smad2/3 are sensors of the interplay between effects of TGF-β and retinoic acid on monocytic and granulocytic differentiation of HL-60 cells. Blood 2003 Jan;101(2):498-507.
- 27. Hata A, Lo RS, Wotton D, Lagna G, Massagué J. Mutations increasing autoinhibition inactivate tumour suppressors Smad2 and Smad4. Nature 1997 Jul;388(6637):82-87.
- 28. Stopa M, Anhuf D, Terstegen L, Gatsios P, Gressner AM, Dooley S. Participation of Smad2, Smad3, and Smad4 in transforming growth factor β (TGF-β)-induced activation of Smad7. THE TGF-β response element of the promoter requires functional Smad binding element and E-box sequences for transcriptional regulation. J Biol Chem 2000 Sep;275(38):29308-29317.
- 29. Xu W, Angelis K, Danielpour D, Haddad MM, Bischof O, Campisi J, et al. Ski acts as a co-repressor with Smad2 and Smad3 to regulate the response to type β transforming growth factor. Proc Natl Acad Sci U S A 2000 May;97(11):5924-5929.
- 30. Huang C, Liu H, Gong X-L, Wu L, Wen B. Expression of DNA methyltransferases and target microRNAs in human tissue samples related to sporadic colorectal cancer. Oncol Rep 2016 Nov;36(5):2705-2714.
- 31. Wang C, Jia Z, Ma H, Cao D, Wu X, Wen S, et al. DNA methyltransferase 3a rs1550117 genetic polymorphism predicts poor survival in gastric cancer patients. Int J Clin Exp Pathol 2015 Nov;8(11):14864-14874.
- 32. Cacan E, Greer SF, Garnett-Benson C. Radiation-induced modulation of immunogenic genes in tumor cells is regulated by both histone deacetylases and DNA methyltransferases. Int J Oncol 2015 Dec;47(6):2264-2275.
- 33. Zhang W, Xu Y, Ma G, Qi W, Gu H, Jiang P. Genetic polymorphism of DNA methyltransferase 3A rs1550117 A> G and risk of cancer: a meta-analysis. J Invest Surg 2015;28(6):346-353.
- 34. Norollahi SE, Alipour M, Rashidy-Pour A, Samadani AA, Larijani LV. Regulatory fluctuation of WNT16 gene expression is associated with human gastric adenocarcinoma. J Gastrointest Cancer 2019 Mar;50(1):42-47.
- 35. Samadani AA, Nikbakhsh N, Taheri H, Shafaee S, Fattahi S, Pilehchian Langroudi M, et al. cdx1/2 and klf5 expression and epigenetic modulation of sonic hedgehog signaling in gastric adenocarcinoma. Pathol Oncol Res 2019 Jul;25(3):1215-1222.
- 36. Pilehchian Langroudi M, Nikbakhsh N, Samadani AA, Fattahi S, Taheri H, Shafaei S, et al. FAT4 hypermethylation and grade dependent downregulation in gastric adenocarcinoma. J Cell Commun Signal 2017 Mar;11(1):69-75.
- 37. Liu X, Luo M, Wu K. Epigenetic interplay of histone modifications and DNA methylation mediated by HDA6. Plant Signal Behav 2012 Jun;7(6):633-635.
- 38. Nowacka-Zawisza M, Wiśnik E. DNA methylation and histone modifications as epigenetic regulation in prostate cancer. Oncol Rep 2017 Nov;38(5):2587-2596.
- 39. Hongwei S, Tiezhu A, Shanhua P, Chunsheng W. Mammalian DNA methylation and its roles during the induced re-programming of somatic cells. Yi chuan= Hereditas 2014;36(5):431-438.
- 40. Juriloff DM, Harris MJ. Hypothesis: the female excess in cranial neural tube defects reflects an epigenetic drag of the inactivating X chromosome on the molecular mechanisms of neural fold elevation. Birth Defects Res A Clin Mol Teratol 2012 Oct;94(10):849-855.
- 41. Lamba G, Zaidi SK, Luebbers K, Verschraegen C, Stein GS, Rosmarin A. Epigenetic landscape of acute myelogenous leukemia–moving toward personalized medicine. J Cell Biochem 2014 Oct;115(10):1669-1672.
- 42. Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov 2006 Jan;5(1):37-50.
- 43. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004 Feb;4(2):143-153.
- 44. Hellebrekers DM, Griffioen AW, van Engeland M. Dual targeting of epigenetic therapy in cancer. Biochimica et Biophysica Acta (BBA) Rev Can 2007;1775(1):76-91.
- 45. Cheung HH, Lee TL, Rennert OM, Chan WY. DNA methylation of cancer genome. Birth Defects Res C Embryo Today 2009 Dec;87(4):335-350.
- 46. Pfeifer GP. Defining driver DNA methylation changes in human cancer. Int J Mol Sci 2018 Apr;19(4):1166.
- 47. Samadani AA, Nikbakhsh N, Pilehchian M, Fattahi S, Akhavan-Niaki H. Epigenetic changes of CDX2 in gastric adenocarcinoma. J Cell Commun Signal 2016 Dec;10(4):267-272.
- 48. Ghadami E, Nikbakhsh N, Fattahi S, Kosari-Monfared M, Ranaee M, Taheri H, et al. Epigenetic alterations of CYLD promoter modulate its expression in gastric adenocarcinoma: A footprint of infections. J Cell Physiol 2019 Apr;234(4):4115-4124.
- 49. Fattahi S, Nikbakhsh N, Taheri H, Ghadami E, Kosari-Monfared M, Amirbozorgi G, et al. Prevalence of multiple infections and the risk of gastric adenocarcinoma development at earlier age. Diagn Microbiol Infect Dis 2018 Sep;92(1):62-68.
- 50. Li J, Jin H, Wang X. Epigenetic biomarkers: potential applications in gastrointestinal cancers. ISRN Gastroenterology 2014;2014.
- 51. Norollahi SE, Hamidian SM, Vahidi S, Babaei K, Samadani AA. Modifications of WNT signaling pathway genes including WNT1, KLF5 and WNT16 in colorectal cancer. Gene Rep 2020;20:100733.
- 52. Rodriguez J, Frigola J, Vendrell E, Risques R-A, Fraga MF, Morales C, et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res 2006 Sep;66(17):8462-9468.
- 53. Akhavan-Niaki H, Samadani AA. DNA methylation and cancer development: molecular mechanism. Cell Biochem Biophys 2013 Nov;67(2):501-513.
- 54. Puccini A, Berger MD, Naseem M, Tokunaga R, Battaglin F, Cao S, et al. Colorectal cancer: epigenetic alterations and their clinical implications. Biochim Biophys Acta Rev Cancer 2017 Dec;1868(2):439-448.
- 55. Boot A, Oosting J, van Eendenburg JD, Kuppen PJ, Morreau H, van Wezel T. Methylation associated transcriptional repression of ELOVL5 in novel colorectal cancer cell lines. PLoS One 2017 Sep;12(9):e0184900.
- 56. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis 2010 Jan;31(1):27-36.
- 57. Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 2008 Jun;9(6):465-476.
- 58. Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007 Feb;128(4):683-692.
- 59. Nejman D, Straussman R, Steinfeld I, Ruvolo M, Roberts D, Yakhini Z, et al. Molecular rules governing de novo methylation in cancer. Cancer Res 2014 Mar;74(5):1475-1483.
- 60. Norollahi SE, Mansour-Ghanaei F, Joukar F, Ghadarjani S, Mojtahedi K, Gharaei Nejad K, et al. Therapeutic approach of Cancer stem cells (CSCs) in gastric adenocarcinoma; DNA methyltransferases enzymes in cancer targeted therapy. Biomed Pharmacother 2019 Jul;115:108958.
- 61. Samadani AA, Norollahi SE, Rashidy-Pour A, Mansour-Ghanaei F, Nemati S, Joukar F, et al. Cancer signaling pathways with a therapeutic approach: An overview in epigenetic regulations of cancer stem cells. Biomed Pharmacother 2018 Dec;108:590-599.
- 62. Samadani AA, Noroollahi SE, Mansour-Ghanaei F, Rashidy-Pour A, Joukar F, Bandegi AR. Fluctuations of epigenetic regulations in human gastric Adenocarcinoma: how does it affect? Biomed Pharmacother 2019 Jan;109:144-156.
- 63. Belshaw NJ, Pal N, Tapp HS, Dainty JR, Lewis MP, Williams MR, et al. Patterns of DNA methylation in individual colonic crypts reveal aging and cancer-related field defects in the morphologically normal mucosa. Carcinogenesis 2010 Jun;31(6):1158-1163.
- 64. Klutstein M, Nejman D, Greenfield R, Cedar H. DNA methylation in cancer and aging. Cancer Res 2016 Jun;76(12):3446-3450.
- 65. Sameer AS, Nissar S. Epigenetics in diagnosis of colorectal cancer. Mol Biol Res Commun 2016 Mar;5(1):49-57.
- 66. Ma Z, Williams M, Cheng YY, Leung WK. Roles of methylated DNA biomarkers in patients with colorectal cancer. Disease Markers 2019;2019.
- 67. deVos T, Molnar B. Screening for colorectal cancer based on the promoter methylation status of the septin 9 gene in plasma cell free DNA. J Clin Epigenetics 2017;3:1-6.
- 68. Danese E, Montagnana M. Epigenetics of colorectal cancer: emerging circulating diagnostic and prognostic biomarkers. Ann Transl Med 2017 Jul;5(13):279.
- 69. McCabe MT, Lee EK, Vertino PM. A multifactorial signature of DNA sequence and polycomb binding predicts aberrant CpG island methylation. Cancer Res 2009 Jan;69(1):282-291.
- 70. Lao VV, Grady WM. Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol 2011 Oct;8(12):686-700.
- 71. Yang SX, Nguyen D, Rubinstein L, Chen AP, Doroshow JH. Abstract B12: DNA methyltransferase 3A expression in cancer cells following treatment with DNMT inhibitors and in human solid tumors. AACR 2015;14(12).
- 72. Freytag SO. Enforced expression of the c-myc oncogene inhibits cell differentiation by precluding entry into a distinct predifferentiation state in G0/G1. Mol Cell Biol 1988 Apr;8(4):1614-1624.
- 73. Prendergast GC, Ziff EB. Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science 1991 Jan;251(4990):186-189.
- 74. Elbadawy M, Usui T, Yamawaki H, Sasaki K. Emerging roles of C-Myc in cancer stem cell-related signaling and resistance to cancer chemotherapy: a potential therapeutic target against colorectal cancer. Int J Mol Sci 2019 May;20(9):2340.
- 75. Sharrard RM, Royds JA, Rogers S, Shorthouse AJ. Patterns of methylation of the c-myc gene in human colorectal cancer progression. Br J Cancer 1992 May;65(5):667-672.
- 76. Wu MY, Hill CS. Tgf-β superfamily signaling in embryonic development and homeostasis. Dev Cell 2009 Mar;16(3):329-343.
- 77. Oshimori N, Fuchs E. The harmonies played by TGF-β in stem cell biology. Cell Stem Cell 2012 Dec;11(6):751-764.
- 78. Gaarenstroom T, Hill CS, eds. TGF-β signaling to chromatin: how Smads regulate transcription during self-renewal and differentiation. Seminars in cell & developmental biology; Elsevier; 2014. p. 107-118.
- 79. Bertero A, Brown S, Madrigal P, Osnato A, Ortmann D, Yiangou L, et al. The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency. Nature 2018 Mar;555(7695):256-259.
- 80. Vieira AT, Fukumori C, Ferreira CM. New insights into therapeutic strategies for gut microbiota modulation in inflammatory diseases. Clin Transl Immunology 2016 Jun;5(6):e87.
- 81. Sana J, Faltejskova P, Svoboda M, Slaby O. Novel classes of non-coding RNAs and cancer. J Transl Med 2012 May;10(1):103.
- 82. Ruan K, Fang X, Ouyang G. MicroRNAs: novel regulators in the hallmarks of human cancer. Cancer Lett 2009 Nov;285(2):116-126.
- 83. Esteller M. Non-coding RNAs in human disease. Nat Rev Genet 2011 Nov;12(12):861-874.