Sanjad-Sakati syndrome is characterized by hypoparathyroidism, retardation, and dysmorphism; it is an autosomal recessive inherited disorder that has only been observed in patients of Arabic origin.1 It was first reported by Sanjad et al.2 Sanjad-Sakati syndrome is typically caused by genetic mutations in the tubulin-folding cofactor E (TBCE; 604934) gene, which is located directly on the 1q42.3 chromosome.3
This disorder is characterized usually by inherent hypoparathyroidism that leads to hypocalcemic seizures of early onset, intrauterine growth retardation craniofacial dysmorphism, and mental retardation. The typical physical appearance includes a narrow, long face, beaked nose, deep-set eyes, large ears, thin lips, long philtrum, and micrognathia.4,5,6,7,8 This case was reported from the Fallujah Teaching Hospital for Women and Children in Fallujah, Iraq.
Case Report
A 35-day-old female patient presented with generalized tonic-clonic seizures but without any history of fever or trauma. Patient investigations showed serum calcium of 1 mmol/L (normal range = 2.3–2.65 mmol/L), alkaline phosphatase of 199 u/L (normal range = 90–180 u/L), phosphorus of 3.05 mmol/L (normal range = 1.4–2.5 mmol/L), and parathyroid hormone of 1.20 ng/L (normal range = 10–65 ng/L). The complete blood count, renal function, liver function test, and urine examination were all within the normal ranges. Abdominal echo ultrasound study was normal. The diagnosis was confirmed by a genetic study through whole exome sequence by CENTOGENE Company in Germany that revealed TBCE, c.155_166del p.(Ser52_Gly55del).
This case was diagnosed as primary hypo-parathyroidism and treated with parenteral anticonvulsants, calcium, and vitamin D supplements. The seizures were then controlled. She was a term baby, delivered vaginally weighing 2 kg at birth, and the fourth offspring of a consanguineous marriage with a history of one abortion.
Length, weight, and head circumference were less than -2 SD from the mean for her age. This patient had typical physical findings [Figure 1] consisting of microcephaly, a narrow and long face, a beaked nose, deep-set eyes, large ears, a long philtrum, a small hand, and small feet. The systemic examination, including the cardiovascular system, was normal.
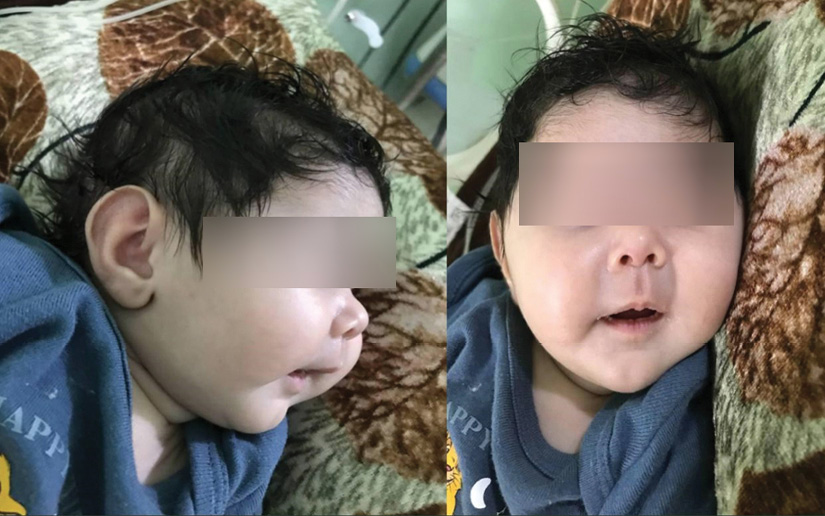 Figure 1: Newborn displaying a beaked nose, large ears, long philtrum, and thin lips.
Figure 1: Newborn displaying a beaked nose, large ears, long philtrum, and thin lips.
Discussion
Sanjad-Sakati syndrome is a very rare inherited autosomal recessive disorder that is distributed equally in both males and females. The gene location of this syndrome is 1q42.3 chromosome.3 This syndrome is characterized by hypoparathyroidism, growth retardation, atypical facial features, and a variable degree of mental retardation. The most common abnormal physical findings of this syndrome are microcephaly, narrow and long face, beaked nose, deep-set eyes, large ears, thin lips, long philtrum, and small hands and feet.4,5,6,7,8 Some of these features resemble Kenny-Caffey syndrome, DiGeorge syndrome, and familial isolated hypoparathyroidism, but the absence of a cardiovascular lesion, lymphocytopenia, or skeletal malformation makes it a different problem.9 Ophthalmological assessments help us to differentiate Sanjad-Sakati and Kenny-Caffey syndromes. Corneal opacity and nanophthalmos are present in Kenny-Caffey syndrome patients, while these features are not described in Sanjad-Sakati syndrome patients apart from deep-set eyes.10
This syndrome is commonly described in people of Arabic origin in the Middle East; the registered cases were from Saudi Arabia, Kuwait, Qatar, Oman, Morocco, and Jordan.11,12,13,14,15 The first reported syndrome was by Sanjad et al,2 then 12 cases were published in 1991;4 six in boys and six in girls. Symptoms also started in the neonatal period for nine patients. All of these cases have hypoparathyroidism, severe hypocalcemia, and hyperphosphatemia. They also had dysmorphic features and growth retardation. There was no congenital heart disease in these cases, and cell-mediated immunity was normal in five patients. Richardson and Kirk reported eight cases, two were brothers. The weight, height, and occipitofrontal circumference in these cases were < -2 SD for their ages.11 In all of these cases, typical facial features were presented. The first case involved relatives from Qatar in the UK, while the others were from the Middle East region.11 A five-year-old Saudi girl presented with undetected parathyroid hormone and subnormal growth hormone. 13 There was a normal renal response to parathyroid hormone, and growth hormone treatment resulted in a marked increase in weight and height. Hypocalcemia was controlled with 1-alpha-cholecalciferol.13 A genetic study revealed that a Moroccan case with Sanjad-Sakati syndrome presented with the Bedouin c.155-166del mutation,14 and six cases from Jordan had the same deletion, which resembles the mutation in our case.15,16 There is no finding in this case different from previously reported cases.
Due to medical difficulties in making the diagnosis, we should have a high index of suspicion in order not to miss such cases. Because consanguineous marriages are common in our society, genetic counseling is crucial to lowering the risk of genetic mutation.
Conclusion
Although Sanjad-Sakati syndrome is an incurable disease, early diagnosis helps with appropriate palliative treatment of the patient and reduces other associated metabolic and electrolyte disturbances. It also allows for genetic research and counseling of the families, especially in our society where consanguinity is high.
Disclosure
The authors declared no conflicts of interest. Written consent was obtained from the patient’s father.
references
- 1. Ahmed HY, Almeshkhas FM, Hasan ZA, Isa HM. Sanjad Sakati syndrome. Bahrain Med Bull 2020;42(1):67-69.
- 2. Sanjad SA, Sakato NA, Abu-Osba YK, Kaddoura R, Milner RD. A new syndrome of congenital hypoparathyroidism, severe growth failure, and dysmorphic features. Arch Dis Child 1991 Feb;66(2):193-196.
- 3. Touati A, Nouri S, Halleb Y, Kmiha S, Mathlouthi J, Tej A, et al. Additional Tunisian patients with Sanjad-Sakati syndrome: a review toward a consensus on diagnostic criteria. Arch Pediatr 2019 Feb;26(2):102-107.
- 4. Alghamdi S. Oral facial manifestations of Sanjad-Sakati syndrome: a literature review. Children (Basel) 2022 Mar;9(4):448.
- 5. Hassona Y, Rajab L, Taimeh D, Scully C. Sanjad-Sakati syndrome: oral health care. Med Princ Pract 2018;27(3):293-296.
- 6. Al-Malik MI. The dentofacial features of Sanjad-Sakati syndrome: a case report. Int J Paediatr Dent 2004 Mar;14(2):136-140.
- 7. Naguib KK, Gouda SA, Elshafey A, Mohammed F, Bastaki L, Azab AS, et al. Sanjad-Sakati syndrome/Kenny-Caffey syndrome type 1: a study of 21 cases in Kuwait. East Mediterr Health J 2009;15(2):345-352.
- 8. AlAyed OA. Sanjad-Sakati syndrome and its association with superior mesenteric artery syndrome. Case Rep Pediatr 2014;2014:108051.
- 9. Bashar M, Taimur M, Amreek F, Sayeed KA, Tahir A. Endocrinological manifestations of Sanjad-Sakati syndrome. Cureus 2020 Jun;12(6):e8770.
- 10. Khan AO, Al-Assiri A, Al-Mesfer S. Ophthalmic features of hypoparathyroidism-retardation-dysmorphism. J AAPOS 2007 Jun;11(3):288-290.
- 11. Richardson RJ, Kirk JM. Short stature, mental retardation, and hypoparathyroidism: a new syndrome. Archives of Disease in Childhood 1990 Oct 1;65(10):1113-1117.
- 12. Kalam MA, Hafeez W. Congenital hypoparathyroidism, seizure, extreme growth failure with developmental delay and dysmorphic features–another case of this new syndrome. Clin Genet 1992 Sep;42(3):110-113.
- 13. Marsden D, Nyhan WL, Sakati NO. Syndrome of hypoparathyroidism, growth hormone deficiency, and multiple minor anomalies. Am J Med Genet 1994 Sep;52(3):334-338.
- 14. Ratbi I, Lyahyai J, Kabiri M, Banouar M, Zerkaoui M, Barkat A, et al. The Bedouin mutation c.155-166del of the TBCE gene in a patient with Sanjad-Sakati syndrome of Moroccan origin. Ann Saudi Med 2015;35(2):170-172.
- 15. Pal K, Moammar H, Mitra DK. Visceral myopathy causing chronic intestinal pseudoobstruction and intestinal failure in a child with Sanjad-Sakati syndrome. J Pediatr Surg 2010 Feb;45(2):430-434.
- 16. Kerkeni E, Sakka R, Sfar S, Bouaziz S, Ghedira N, Ben Ameur K, et al. Sanjad-Sakati syndrome in a Tunisian child. Arch Pediatr 2015 Sep;22(9):951-955.