Despite being a rare pediatric disease with a prevalence of about one per 20 000 live births, biliary atresia (BA) is the most frequent reason for surgery in infants with persistent jaundice and the leading cause of pediatric liver transplants (LTs).1,2 BA is an idiopathic inflammatory liver disease that manifests in the neonatal period, and characterized by progressive fibrosis and obstruction of both intrahepatic and extrahepatic bile ducts.3 It presents with symptoms of cholestatic jaundice, which, if left untreated, results in cirrhosis and death in 50–80% of patients within a year and 90–100% at the age of three.4,5
Currently, Kasai hepatic portoenterostomy (HPE)—commonly known as Kasai procedure—and LT are the only two effective treatments for BA. HPE is the primary treatment, performed as soon as the diagnosis of BA is confirmed; LT is usually indicated if there is inadequate bile clearance after the Kasai procedure, growth failure, progressive liver dysfunction, or subsequent decompensated cirrhosis.6,7
A well-performed Kasai procedure can transform BA from a fatal disease to a successfully treated one, in addition to delaying the requirement of LT. However, LT may become inevitable for around 60–80% of patients by adulthood due to the emergence of unmanageable complications.8–10 A systematic review on the outcomes in 184 adult patients with BA who survived to adulthood (> 20 years) with their native livers found that 60.5% of them ultimately had significant complications such as bacterial cholangitis, portal hypertension and its sequelae, and hepatocellular carcinoma.10 These figures are supported by other studies.11
Overall survival rates and quality of life have improved impressively over the last few decades for those who successfully went through both Kasai and LT, but potential contraindications to surgery and accessibility of liver graft continue to impact the success rate. In the Middle East, only a few LT centers are available. Cultural and religious influences that discourage organ donation and low public awareness have also impacted the transplantation rates in this region.12,13
Studies tackling BA in the Middle East are scarce. Therefore, our study aimed to assess the prevalence of BA in Bahrain, to review the clinical presentations, diagnoses, and management including surgical interventions. We also aimed to evaluate the long-term survival rates and other outcomes of BA and analyze the factors that affected the survival of the patients encountered in our center.
Methods
In this retrospective cohort study, the subjects were all infants diagnosed with BA or followed-up for BA at Salmaniya Medical Complex, Manama, Bahrain, between January 1993 and December 2021.
Diagnosis of BA for each patient was observed to have been confirmed using a combination of radiographic and liver histology results. Not all patients included in our study had the Kasai procedure in Bahrain. All Kasai procedures performed in Bahrain were open surgeries. Those who required LT had been referred overseas due to the non-availability of the facility in the country.
Data retrieved from the hospital database included the year of presentation, age, sex, nationality, gestational age, mode of delivery, birth weight, perinatal history, parental consanguinity, family history of liver disease or BA, age at admission, age at diagnosis, the onset of jaundice, presence of dark urine, clay-colored stool, pruritus, vomiting, fever, and associated conditions. Physical examination findings including admission weight, abdominal distension, hepatomegaly, splenomegaly, ophthalmological findings, and involvement of other systems were also collected.
Laboratory, radiological, and histopathology data reviewed included the results of hemoglobin level, platelet count, coagulation profile, serum total protein, serum albumin and globulin, total bilirubin, direct and indirect bilirubin levels, alkaline phosphatase, alanine aminotransferase, gamma-glutamyl transferase, serum ammonia, serum lactate, metabolic workup, glucose, cholesterol and triglyceride, serum iron and ferritin level, serum alpha-1 antitrypsin level, sweat chloride test, and thyroid function tests. The results of urine culture, combined serology of toxoplasma, rubella, cytomegalovirus, and herpes simplex virus were also reviewed. Findings of chest X-ray, abdominal ultrasound (US), echocardiography, hepatobiliary iminodiacetic acid (HIDA) scan, liver biopsy and age at liver biopsy were collected. US scans with triangular cord signs, contracted gallbladder, and non-visualized common bile duct were taken as significant positive indicators for BA. A positive HIDA scan was considered to indicate that there was no uptake or excretion into the bowel. The infant’s age at Kasai procedure and direct bilirubin levels before and after the procedure were also noted.
The patient outcomes were divided into three scenarios: (1) survival with complications, (2) death, and (3) need for LT. Age at LT, age at death, and cause of death were noted. Total serum bilirubin and age at the last follow-up visit were also acquired.
The collected data were analyzed using IBM SPSS Statistics (IBM Corp. Released 2012. IBM SPSS Statistics for Windows, Version 21.0. Armonk, NY: IBM Corp.). The frequencies, percentages, and overall prevalence of BA in Bahrain were calculated. Group data were presented as a mean±SD for normally distributed variables or median and interquartile range (IQR) for non-normally distributed variables. For normally distributed continuous variables, the independent student t-test was used to compare the group means, while the Mann-Whitney U test was used to compare group medians for non-normally distributed continuous variables including the difference in prevalence between the Bahraini and non-Bahraini patients. Chi-square and Fisher's exact tests were used to compare categorical variables. P-values < 0.05 were considered statistically significant. Survival analysis based on procedure type was estimated via the Kaplan-Meier method.
This study followed the Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans and was ethically approved by the Secondary Health Care Research Committee, Salmaniya Medical Complex, Government Hospitals, Bahrain (IRB number: 103020821). It was noted that all parents/guardians had signed their written informed consent upon admission of their wards to the hospital.
Results
During the study period, 27 infants were confirmed to have BA. One of them was excluded from our study due to a lack of information on the outcome. Thus, 26 infants comprised our study population. This included one Bahraini female initially diagnosed with Alagille syndrome in whom BA was confirmed intraoperatively during the Kasai procedure.
Demographic data of the patients with BA are shown in Table 1. Majority (15; 57.7%) were females. Sixteen (61.5%) infants were Bahraini and the remaining 10 (38.5%) were non-Bahraini, comprising three Egyptians (11.5%), three Pakistanis (11.5%), two Syrians (7.7%), one Indian (3.8%), and one Yemeni (3.8%). Clinical signs and physical examination findings are shown in Table 2. Most patients had clay-colored stool and hepatomegaly. According to the 2019 national health statistics, the total population of Bahrain was 1 483 756. The pediatric population was 378 410 (25.5%), of whom 280 029 (74.0%) were Bahraini and 98 381 (26.0%) non-Bahraini. The overall prevalence of BA was 6.87 per 100 000 (0.007%) of the pediatric population. The prevalence was significantly higher among Bahraini than non-Bahraini children, at 5.7 per 100 000 (0.006%) and 1.02 per 100 000 (0.001%), respectively (p < 0.001).
Table 1: Demographic data of 26 infants with biliary atresia.
|
Sex
|
|
|
Female
|
15 (57.7)
|
|
Male
|
11 (42.3)
|
|
Nationality
|
|
Bahrain
|
16 (61.5)
|
|
Non-Bahraini
|
10 (38.5)
|
|
Gestational age (n = 22)
|
|
Term
|
19 (90.5)
|
|
Preterm
|
2 (9.5)
|
|
Type of delivery (n = 21)
|
|
Normal
|
17 (85.0)
|
|
Cesarean section
|
3.0 (15.0)
|
|
Positive perinatal history† (n = 21)
|
2 (9.5)
|
|
Parental consanguinity (n = 19)
|
5 (26.3)
|
|
Age at admission (days), median, IQR (n = 21)
|
64 (32–124)
|
|
Age at diagnosis (days), median, IQR (n = 19)
|
56 (30–90)
|
|
Age at time of study (year), median, IQR
|
10.3 (7.2–17.2)
|
|
Admission weight (kg), mean ± SD (n = 11)
|
4.8 ± 1.4
|
†One mother had gestational diabetes and another mother was a hepatitis B carrier; IQR: interquartile range.
Table 2: Clinical signs and physical examination of 22 infants with biliary atresia.
|
Clinical signs
|
|
|
Clay stool
|
19 (86.4)
|
|
Dark urine
|
18 (81.8)
|
|
Pruritus
|
3.0 (13.6)
|
|
Vomiting
|
1 (4.5)
|
|
Fever
|
1 (4.5)
|
|
Associated syndromes*
|
1 (4.5)
|
|
Physical examination
|
|
|
Hepatomegaly (n = 21)
|
19 (90.5)
|
|
Splenomegaly (n = 21)
|
6 (28.9)
|
|
Abdominal distension
|
6 (27.3)
|
|
Heart murmur
|
3 (13.6)
|
*One infant had Alagille syndrome; ‡posterior embryotoxon in one patient.
Results of antenatal US were available for 21 (80.8%) mothers and all were reported to be normal. Heart murmurs were noticed in three (13.6%) infants during physical examination at the presentation. Echocardiography revealed peripheral pulmonary stenosis in association with Alagille syndrome, ventricular septal defect, and patent ductus arteriosus in each patient. The median age at the onset of jaundice was five (IQR = 1–30) days. The median age at diagnosis was 56 (IQR = 30–90) days.
All the patients had direct hyperbilirubinemia at presentation with elevated levels of serum direct bilirubin (151.0±68.0 µmol/L; normal = 0–5); alkaline phosphatase (740.0±270.0 IU/L; normal = 150–420), alanine aminotransferase (142.0±77.0 IU/L; normal: < 33), and gamma-glutamyl transferase (791.0±778.0 IU/L; normal < 185). Three out of 14 infants (21.4%) were cytomegalovirus positive on toxoplasma, rubella, cytomegalovirus, and herpes screening, and two out of 12 (16.7%) patients had positive urine cultures for Enterobacter/Klebsiella and Escherichia coli.
Abdominal US yielded positive findings in 11 of 14 (78.6%) patients, while HIDA scan results were positive for all the 12 (100%) patients who were subjected to it. Liver biopsy confirmed BA in all the 18 (100%) cases tested for the same.
Patients management and outcomes are displayed in Table 3. The Kasai procedure was performed in 23 (88.5%) infants at a median age of 61 (IQR = 56–90) days. Three (11.5%) patients underwent primary LT at 4.5, 7.3, and 30.0 months, respectively. One patient had a Kasai revision two weeks after the initial Kasai procedure. Seventeen of 23 (73.9%) infants had the Kasai procedure in Bahrain, three (13.0%) in Egypt, two (8.7%) in Pakistan, and one (4.3%) in Jordan. Thirteen (50.0%) patients had the Kasai procedure alone, while the remaining ten (38.5%) required LT after the Kasai procedure at a median age of 0.9 years (IQR = 0.8–7.0). For the ten infants who underwent both Kasai and transplant, the median time period between the two procedures was 0.8 years (IQR = 0.6–7.0); the youngest patient's age at transplant was 0.4 years, and the oldest was 13 years. All liver transplantation procedures were performed abroad—seven (53.8%) in Saudi Arabia, three (23.1%) in India, and one (7.7%) each in Turkey, Egypt, and the UK. All received liver tissue from living donors. Donor information was available for 12 patients, with mothers accounting for seven (58.3%), fathers for four (33.3%), and an uncle for one (8.3%). As of December 2021 (the end of our data collection period), 10 of 13 (76.9%) LT recipients remained alive and they were continuing to receive the immunosuppressant, tacrolimus.
Table 3: Survival characteristics in infants with biliary atresia.
|
Kasai procedure alone
|
13
|
6
|
7
|
53.8
|
11.2 (6.26–15.77)
|
6.4 ± 5.3
|
0.422
|
|
Kasai and liver transplant
|
10
|
2
|
8
|
80.0
|
23.1 (16.64–29.56)
|
10.7 ± 8.1
|
|
|
Primary liver transplant
|
3
|
1
|
2
|
66.7
|
10.9 (2.25–19.54)
|
5.8 ± 9.0
|
|
*Follow-up time is measured from the initial treatment date i.e., Kasai or liver transplant until the event (death) occurs or study end date; †95% CI.
Cirrhosis (16 cases; 76.2%) and cholangitis (13 cases; 61.9%) were the two most frequent complications that our patients experienced prior to LT. Other complications included portal hypertension in six (28.6%) patients, ascites in four (19.0%), esophageal varices in three (14.3%), coagulopathy in two (9.5%), and intestinal obstruction in two (9.5%) patients. In total, 13 (54.2%) transplants were performed due to reasons such as late presentation, failed Kasai, end-stage liver failure, recurrent cholangitis, and other complications.
Four BA patients have entered adulthood, and the oldest was aged 28 years. Three of them have had LT, including the eldest patient. They had no complications related to the transplant or immunosuppressive medications. The fourth patient was approaching the age of 20 years with her native liver but is currently being reviewed for LT because of recurrent cholangitis.
The overall mortality rate during the study period was 34.6% (9 of 26 patients). The median age at death was 1.6 years (IQR = 0.9–3.5). Five were female, and four were male. One of the patients who underwent primary LT aged seven months died after a month due to graft rejection. Six of 13 (46.2%) patients who had only the Kasai procedure died awaiting LT donors. In one infant, Kasai was performed at the age of six months but later died aged two years owing to progressive liver failure. Two (20.0%) of the ten patients who underwent both Kasai and LT died because of multiorgan failure.
The native liver survival (NLS) rate was 53.8% and the overall liver survival (OLS) rate was 65.4% [Figure 1]. The survival rate reached up to 80% in those who underwent both Kasai and LT. The mean follow-up time measured from the initial treatment date i.e., from Kasai/LT until death or the end of our data collection period (December 2021) was 8.0±7.0 years. Data available for 15 (57.7%) patients revealed median total serum bilirubin to be 13 µmol/L (IQR = 8–48) at the last follow-up.
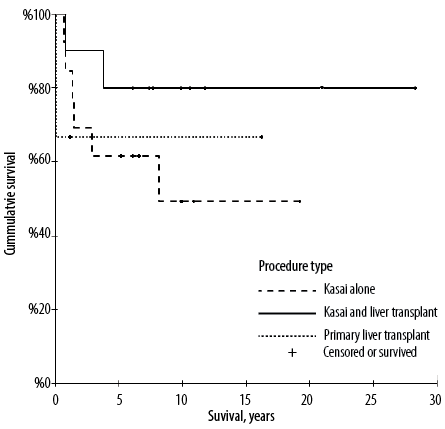 Figure 1: Survival analysis in patients with biliary atresia according to procedure type using the Kaplan-Meier method.
Figure 1: Survival analysis in patients with biliary atresia according to procedure type using the Kaplan-Meier method.
Among demographic data, birth weight and age at diagnosis seemed to have a predictive effect on survival [Table 4]. Infants with lower mean birth weight and older mean age at diagnosis were found to be associated with an increased number of survivors (p < 0.029 and p < 0.044, respectively). After excluding one outlier patient who was diagnosed very late at the age of 231 days, the mean age at diagnosis was 36.6±18.8 days for dead patients compared to 65.9±28.7 days for survived patients (p = 0.053). None of the other factors such as sex, nationality, type of delivery, gestational age, consanguinity, age at Kasai, age at LT, and complications, had a statistically significant impact on survival.
Table 4: Predictors of survival in patients with biliary atresia.
|
Sex
|
|
Female
|
10 (66.7)
|
5 (33.3)
|
0.873‡
|
|
Male
|
7.0 (63.6)
|
4 (36.4)
|
|
|
Nationality
|
|
Bahraini
|
10 (62.5)
|
6 (37.5)
|
0.696‡
|
|
Non-Bahraini
|
7 (70.0)
|
3 (30.0)
|
|
|
Gestational age (n = 22)
|
|
Term
|
16 (80.0)
|
4 (20.0)
|
0.334‡
|
|
Preterm
|
1 (50.0)
|
1 (50.0)
|
|
Type of delivery (n = 21)
|
|
Normal
|
13 (76.5)
|
4 (23.5)
|
0.950‡
|
|
Caesarean section
|
3 (75.0)
|
1 (25.0)
|
|
Consanguinity (n = 5)
|
4 (80.0)
|
1 (20.0)
|
0.709‡
|
|
Birth weight (kg), mean ± SD (n = 22)
|
16 (2.9 ± 0.5)
|
6(3.5 ± 0.5)
|
0.029§ (-1.10–-0.07)
|
|
Admission weight (kg), mean ± SD (n = 11)
|
7 (5.2 ± 1.6)
|
4 (4.1 ± 0.8)
|
0.225§ (-0.81–2.99)
|
|
Age at admission (day), mean ± SD (n = 21)
|
15 (170.0 ± 307.0)
|
6 (43.0 ± 47.0)
|
0.080**
|
|
Age at diagnosis (day), mean ± SD (n = 19)
|
14 (76.0 ± 49.0)
|
5 (37.0 ± 19.0)
|
0.044**
|
|
Age at Kasai (day), mean ± SD (n = 23)
|
15 (73.0 ± 31.0)
|
8 (78.0 ± 44.0)
|
0.591**
|
|
Age at liver transplant (year), mean ± SD (n = 13)
|
10 (4.0 ± 4.3)
|
3 (0.7 ± 0.2)
|
0.077**
|
|
Recurrent cholangitis (n = 13)
|
9 (69.2)
|
4 (30.8)
|
0.68‡
|
|
Ascites (n = 4)
|
1 (25.0)
|
3 (75.0)
|
0.065‡
|
‡Fischer’s exact test; §Student’s t-test; **Mann-Whitney U test.
Discussion
This is the first study from Bahrain that evaluated the prevalence, clinical presentations, surgical interventions, and outcomes in patients diagnosed with BA. The prevalence of BA in the Middle East is low compared to Asia-Pacific and Europe. In a Saudi Arabian study, BA amounted to 4.7% of the causes of infantile cholestasis, significantly lower the range of 20–25% reported from LT centers in Western countries, which also placed BA as the second most common indication for LT (after progressive familial intrahepatic cholestasis).14
In this study, the majority of BA patients were females (57.7%). Female predominance has also been reported in other studies from the Middle East,15–17 East Asia,18,19 and Western countries.20–22 This female predominance might have a scientific explanation. One of the many hypotheses for the cause of BA is that viral infection of cholangiocytes leads to an exaggerated humoral autoimmune response which eventually results in progressive bile duct damage and fibrosis. Anti-alpha-enolase antibodies have also been discovered in BA which is typically found in patients with autoimmune hepatitis.23–25 This supports the autoimmune hypothesis of the etiology of BA, as it has previously been established that autoimmune diseases are more prevalent in females than males.
Prompt diagnosis of BA and its timely correction depends on early referral, which in turn depends on vigilant caregivers recognizing symptoms and reporting them. The median age at diagnosis in our sample was 56 days and most (86.4%) infants presented with clay-colored stools, a characteristic of BA. In Taiwan, an infant stool color card (SCC) is being used as a nationwide screening program since 2004 which has resulted in early detections of BA, enabling earlier Kasai operations.26 This scheme was also adopted in Japan where a large-scale study conducted a mass screening program over a period of 19 years and evaluated the sensitivity and specificity of the SCC.27 The sensitivity and specificity of the cards at one-month health check up were 76.5% (95% CI: 62.2–90.7), and 99.9% (95% CI: 99.9–100.0), respectively.27 Patients who reported stool color indicative of BA were more likely to have Kasai performed within 60 days. El-Shabrawi et al,28 implemented SCC in tertiary centers in Egypt and the card proved to be highly sensitive (93.48%; 95% CI: 82.1–98.63) and specific (90.32%; 95% CI 80.12–96.37) for early diagnosis of BA, even where parental education level was low. Perhaps employing and promoting measures such as stool cards and measurement of serum conjugated bilirubin as part of newborn screening at our center may expedite earlier diagnosis of BA in children and facilitate better outcomes.29
Conflicting results seen in our study, where patients diagnosed at an older age and with lower mean birth weight happened to be related to more survivors, might be due to the small sample of 26 babies in addition to missing data and because we did not find this reported elsewhere.
The median age for the Kasai procedure in our sample was 61 days which was comparable to other cohorts as shown in Table 5.5,8,16–18 There is a consensus between multiple studies that the Kasai procedure performed at earlier ages is associated with favorable outcomes with better jaundice clearance.1,5,18,35,36 The optimal age for infants to undergo the Kasai procedure has not yet been identified but ≤ 60 days has been suggested.35,36 Okubo et al,36 classified BA patients into subgroups: early Kasai ≤ 60 days and late Kasai > 60 days. The early Kasai group was further subdivided into ≤ 30, 31–45, and 46–60 days. Outcome analysis revealed that jaundice clearance rates and long-term survival were higher in the early Kasai group. The best results were seen in infants who had surgery during ≤ 30 and 46–60 days. Curiously, babies aged 31–45 days did not fare as well.
Table 5: Review of treatments and survival outcomes of patients with biliary atresia in studies published after 2010.
|
Middle East
|
|
|
|
|
|
|
|
|
|
Bahrain (present study)
|
Isa et al, 2023
|
1993–2021
|
26
|
11:15
|
HPE: 23, LT: 3
|
61
|
5 (53.8)
|
5 (65.4)
|
|
Saudi Arabia
|
Holdar et al,14 2019
|
2008–2015
|
21
|
11:10
|
HPE: 12, LT: 9
|
73
|
4 (14.3)
|
4 (81)
|
|
Saudi Arabia
|
Khayat et al,15 2021
|
1999–2017
|
23
|
10:13
|
HPE
|
77
|
(56)
|
(69.5)
|
|
Iran
|
Naghashi et al,16 2020
|
2006–2017
|
90
|
44:50
|
HPE: 71
|
64.7†
|
5 (40)
|
NR‡
|
|
Egypt
|
Gad et al,17 2021
|
2000–2019
|
410
|
186:224
|
HPE
|
65
|
5 (49.8)
|
5 (51.5)
|
|
Asia-Pacific
|
|
|
|
|
|
|
|
|
|
Hong Kong
|
Chung et al,18 2021
|
1980–2017
|
231
|
97:134
|
HPE
|
59
|
10 (70.7)
|
NR
|
|
China
|
Wang et al,30 2019
|
2009–2012
|
139
|
77:62
|
HPE
|
78.3†
|
5 (58)
|
NR
|
|
India
|
Redkar et al,31 2017
|
2000–2014
|
121
|
75:46
|
HPE
|
96
|
1 (43.8)
|
NR
|
|
Europe
|
|
|
|
|
|
|
|
|
|
Italy
|
Parolini et al,32 2019
|
1975–1996
|
174
|
91:83
|
HPE
|
60
|
5 (41)
|
NR
|
|
Nordic countries
|
Pakarinen et al,20 2018
|
2005–2016
|
154
|
71:83
|
HPE: 148, LT: 6
|
64
|
5 (53)
|
5 (88)
|
|
Netherlands
|
de Vries et al,5 2012
|
1987–2008
|
214
|
110:104
|
HPE
|
59
|
4 (46 ± 4)
|
4 (73 ± 3)
|
|
France
|
Chardot et al,8 2013
|
1986–2009
|
1107
|
NR
|
HPE: 1044
|
59
|
5 (40)
|
5 (81)
|
|
England and Wales
|
Davenport et al,33 2011
|
1999–2009
|
443
|
NR
|
HPE: 424, LT: 15
|
54
|
5 (46)
|
5 (90)
|
|
Africa
|
|
|
|
|
|
|
|
|
†mean days; ‡no record; HPE: hepatic portoenterostomy (Kasai procedure); M:F: male:female; LT: liver transplant.
Post-transplant and waitlist mortality were significantly higher in patients who underwent transplants in infancy compared to those who got transplanted in childhood.33,37 This might be attributed to the smaller stature and lower weight of the former.38 Our series clearly shows that the majority of the children (six out of nine) who passed away did so while waiting for LT and three who died post-transplant were operated on before the age of one year, owing to multiorgan failure and acute graft rejection.
Pre-transplant care is as crucial as post-transplant care. A study published in 2021 analyzed the waitlist mortality determinants in children with BA listed for LT and learned that small size (< 10 kg), raised total bilirubin level, ventilator dependence, and presence of complications such as portal vein thrombosis and ascites at the time of listing were linked with a significantly increased risk for negative outcomes.38 Considering these factors might assist in prioritizing children awaiting LT. Furthermore, maintaining adequate nutrition and avoiding complications during the waiting period is cardinal to successful LT and better outcome.7
In the present study, the Kasai procedure was performed in 23 of 26 (88.5%) infants, one of whom also underwent a Kasai revision surgery. A Kasai revision can be considered in patients with persistent jaundice and multiple episodes of cholangitis. Studies have suggested that revision of Kasai could help defer progressive liver failure and LT, whether or not the initial Kasai was successful.39,40 Conversely, another study cautioned that a Kasai revision should only be performed in patients who did not achieve jaundice-free status after the initial one and that it may increase the risk of morbidity post-LT.41 Despite the contradictory results, it might be worthwhile to consider Kasai revision as an alternative for children waiting for LT.
The five-year NLS rate for BA patients referred to our center was 53.8% which matches most of the other cohorts’ survival rates ranging from 40%–60% [Table 5].8,15–17,20,30–34 Post-transplant survival rate was 76.9% (10/13). Our OLS rate was similar to the cohorts in other Middle Eastern countries but lower in comparison to European or Asian nations. Reasons for such suboptimal outcomes might be attributed to the lack of standardization in surgical and management practices of patients as the Kasai procedure and the LT had been performed in different countries.
The limitations of our study included its retrospective nature, the fact that it was a single-center study, the relatively small cohort, and the lack of standardized treatment. Information regarding different subtypes of BA and other data for a few patients could not be gathered due to missing data. Despite these limitations, our findings fill a long-standing research gap, being the first study from Bahrain focused on BA. Given that BA is one of the most common indications for pediatric liver transplantation, and the catastrophic consequences of delayed diagnosis and treatment of this condition, any new finding on BA in the Middle East is of great relevance. Moreover, the findings of this study can increase awareness about BA among healthcare providers and could form a foundation for any future research, systematic review, or meta-analysis that should include both local and international data.
Conclusion
Although BA is uncommon in infants in Bahrain, it has a disproportionately large role in causing morbidity and mortality despite the available treatment options, namely, HPE and liver transplantation. Our study cohort had relatively low rates of NLS (53.8%) and OLS (65.4%). Most diagnosed patients presented with clay-colored stools—a key warning sign of BA. Thus, implementing an infant stool card may be beneficial in raising parental awareness to facilitate early detection. Further research into the etiology and course of BA and developing novel therapeutic strategies will help improve the survival rates of this rare but debilitating disease.
Disclosure
The authors declared no conflicts of interest. No funding was received for this study.
Acknowledgments
The authors gratefully acknowledge all health personnel of the pediatric department and pediatric surgery division at Salmaniya Medical Complex, Bahrain, involved in the care of children with biliary atresia.
references
- 1. Wildhaber BE. Biliary atresia: 50 years after the first Kasai. ISRN Surg 2012;2012:132089.
- 2. McLin VA. Biliary atresia in adolescents and young adults. Clin Liver Dis (Hoboken) 2013 Oct;2(5):197-199.
- 3. Hartley JL, Davenport M, Kelly DA. Biliary atresia. Lancet 2009 Nov;374(9702):1704-1713.
- 4. Sokol RJ, Mack C, Narkewicz MR, Karrer FM. Pathogenesis and outcome of biliary atresia: current concepts. J Pediatr Gastroenterol Nutr 2003 Jul;37(1):4-21.
- 5. de Vries W, de Langen ZJ, Groen H, Scheenstra R, Peeters PM, Hulscher JB, et al; Netherlands Study Group of Biliary Atresia and Registry (NeSBAR). Biliary atresia in the Netherlands: outcome of patients diagnosed between 1987 and 2008. J Pediatr 2012 Apr;160(4):638-644.e2.
- 6. Squires RH, Ng V, Romero R, Ekong U, Hardikar W, Emre S, et al. Evaluation of the pediatric patient for liver transplantation: 2014 practice guideline by the American association for the study of liver diseases, American society of transplantation and the North American society for pediatric gastroenterology, hepatology and nutrition. Hepatology 2014 Jul;60(1):362-398.
- 7. Sundaram SS, Mack CL, Feldman AG, Sokol RJ. Biliary atresia: indications and timing of liver transplantation and optimization of pretransplant care. Liver Transpl 2017 Jan;23(1):96-109.
- 8. Chardot C, Buet C, Serinet M-O, Golmard J-L, Lachaux A, Roquelaure B, et al. Improving outcomes of biliary atresia: French national series 1986-2009. J Hepatol 2013 Jun;58(6):1209-1217.
- 9. Li S, Ma N, Meng X, Zhang W, Sun C, Dong C, et al. The effects of Kasai procedure on living donor liver transplantation for children with biliary atresia. J Pediatr Surg 2019 Jul;54(7):1436-1439.
- 10. Bijl EJ, Bharwani KD, Houwen RH, de Man RA. The long-term outcome of the Kasai operation in patients with biliary atresia: a systematic review. Neth J Med 2013 May;71(4):170-173.
- 11. Ryon EL, Parreco JP, Sussman MS, Quiroz HJ, Willobee BA, Perez EA, et al. Drivers of hospital readmission and early liver transplant after Kasai portoenterostomy. J Surg Res 2020 Dec;256:48-55.
- 12. Mekkodathil A, El-Menyar A, Sathian B, Singh R, Al-Thani H. Knowledge and willingness for organ donation in the Middle Eastern region: a meta-analysis. J Relig Health 2020 Aug;59(4):1810-1823.
- 13. Khder MA, AlNoaimi LK. Organ donation in Bahrain. Transpl Res Risk Manag 2019;11:59.
- 14. Holdar S, Alsaleem B, Asery A, Al-Hussaini A. Outcome of biliary atresia among Saudi children: a tertiary care center experience. Saudi J Gastroenterol 2019 May-Jun;25(3):176-180.
- 15. Khayat A, Alamri AM, Saadah OI. Outcomes of late Kasai portoenterostomy in biliary atresia: a single-center experience. J Int Med Res 2021 May;49(5):3000605211012596.
- 16. Naghashi S, Rafeey M, Ghojazadeh M, Nikniaz Z, Aslanabadi S, Shoaran M. Biliary atresia in Northwest Iran: epidemiologic features and long-term outcome. Iran J Pediatr 2020;30(2):e96074.
- 17. Gad EH, Kamel Y, Salem TA, Ali MA, Sallam AN. Short- and long-term outcomes after Kasai operation for type III biliary atresia: twenty years of experience in a single tertiary Egyptian center-a retrospective cohort study. Ann Med Surg (Lond) 2021 Jan;62:302-314.
- 18. Chung PH, Chan EK, Yeung F, Chan AC, Mou JW, Lee KH, et al. Life long follow up and management strategies of patients living with native livers after Kasai portoenterostomy. Sci Rep 2021 May;11(1):11207.
- 19. Nio M, Ohi R, Miyano T, Saeki M, Shiraki K, Tanaka K; Japanese Biliary Atresia Registry. Five- and 10-year survival rates after surgery for biliary atresia: a report from the Japanese biliary atresia registry. J Pediatr Surg 2003 Jul;38(7):997-1000.
- 20. Pakarinen MP, Johansen LS, Svensson JF, Bjørnland K, Gatzinsky V, Stenström P, et al; Nordic Pediatric Surgery Study Consortium. Outcomes of biliary atresia in the Nordic countries - a multicenter study of 158 patients during 2005-2016. J Pediatr Surg 2018 Aug;53(8):1509-1515.
- 21. Kumagi T, Drenth JP, Guttman O, Ng V, Lilly L, Therapondos G, et al. Biliary atresia and survival into adulthood without transplantation: a collaborative multicentre clinic review. Liver Int 2012 Mar;32(3):510-518.
- 22. Ng VL, Haber BH, Magee JC, Miethke A, Murray KF, Michail S, et al; Childhood Liver Disease Research and Education Network (CHiLDREN). Medical status of 219 children with biliary atresia surviving long-term with their native livers: results from a North American multicenter consortium. J Pediatr 2014 Sep;165(3):539-546.e2.
- 23. Petersen C, Davenport M. Aetiology of biliary atresia: what is actually known? Orphanet J Rare Dis 2013 Aug;8(1):128.
- 24. Vij M, Rela M. Biliary atresia: pathology, etiology and pathogenesis. Future Sci OA 2020 Mar;6(5):FSO466.
- 25. Mack CL. What causes biliary atresia? Unique aspects of the neonatal immune system provide clues to disease pathogenesis. Cell Mol Gastroenterol Hepatol 2015 May;1(3):267-274.
- 26. Lien T-H, Chang M-H, Wu J-F, Chen H-L, Lee H-C, Chen A-C, et al; Taiwan Infant Stool Color Card Study Group. Effects of the infant stool color card screening program on 5-year outcome of biliary atresia in Taiwan. Hepatology 2011 Jan;53(1):202-208.
- 27. Gu Y-H, Yokoyama K, Mizuta K, Tsuchioka T, Kudo T, Sasaki H, et al. Stool color card screening for early detection of biliary atresia and long-term native liver survival: a 19-year cohort study in Japan. J Pediatr 2015 Apr;166(4):897-902.e1.
- 28. El-Shabrawi MH, Baroudy SR, Hassanin FS, Farag AE. A pilot study of the value of a stool color card as a diagnostic tool for extrahepatic biliary atresia at a single tertiary referral center in a low/middle income country. Arab J Gastroenterol 2021 Mar;22(1):61-65.
- 29. Wang KS; Section on Surgery; Committee on Fetus and Newborn; Childhood Liver Disease Research Network. Newborn screening for biliary atresia. Pediatrics 2015 Dec;136(6):e1663-e1669.
- 30. Wang Z, Chen Y, Peng C, Pang W, Zhang T, Wu D, et al. Five-year native liver survival analysis in biliary atresia from a single large Chinese center: the death/liver transplantation hazard change and the importance of rapid early clearance of jaundice. J Pediatr Surg 2019 Aug;54(8):1680-1685.
- 31. Redkar R, Karkera PJ, Raj V, Bangar A, Hathiramani V, Krishnan J. Outcome of biliary atresia after Kasai’s portoenterostomy: a 15-year experience. Indian Pediatr 2017 Apr;54(4):291-294.
- 32. Parolini F, Boroni G, Milianti S, Tonegatti L, Armellini A, Garcia Magne M, et al. Biliary atresia: 20-40-year follow-up with native liver in an Italian centre. J Pediatr Surg 2019 Jul;54(7):1440-1444.
- 33. Davenport M, Ong E, Sharif K, Alizai N, McClean P, Hadzic N, et al. Biliary atresia in England and Wales: results of centralization and new benchmark. J Pediatr Surg 2011 Sep;46(9):1689-1694.
- 34. De Maayer T, Lala SG, Loveland J, Okudo G, Mohanlal R, Hajinicolaou C. Outcomes of Kasai hepatoportoenterostomy in children with biliary atresia in Johannesburg, South Africa. S Afr Med J 2017 Nov;107(10):12131.
- 35. Jimenez-Rivera C, Jolin-Dahel KS, Fortinsky KJ, Gozdyra P, Benchimol EI. International incidence and outcomes of biliary atresia. J Pediatr Gastroenterol Nutr 2013 Apr;56(4):344-354.
- 36. Okubo R, Nio M, Sasaki H, Japanese Biliary Atresia Society. Impacts of early Kasai portoenterostomy on short-term and long-term outcomes of biliary atresia. Hepatol Commun 2020 Nov;5(2):234-243.
- 37. Arnon R, Annunziato RA, D’Amelio G, Chu J, Shneider BL. Liver transplantation for biliary atresia: is there a difference in outcome for infants? J Pediatr Gastroenterol Nutr 2016 Feb;62(2):220-225.
- 38. Ziogas IA, Ye F, Zhao Z, Cao S, Rauf MA, Izzy M, et al. Mortality determinants in children with biliary atresia awaiting liver transplantation. J Pediatr 2021 Jan;228:177-182.
- 39. Sumida W, Uchida H, Tanaka Y, Tainaka T, Shirota C, Murase N, et al. Review of redo-Kasai portoenterostomy for biliary atresia in the transition to the liver transplantation era. Nagoya J Med Sci 2017 Aug;79(3):415-420.
- 40. Bondoc AJ, Taylor JA, Alonso MH, Nathan JD, Wang Y, Balistreri WF, et al. The beneficial impact of revision of Kasai portoenterostomy for biliary atresia: an institutional study. Ann Surg 2012 Mar;255(3):570-576.
- 41. Shirota C, Uchida H, Ono Y, Murase N, Tainaka T, Yokota K, et al. Long-term outcomes after revision of Kasai portoenterostomy for biliary atresia. J Hepatobiliary Pancreat Sci 2016 Nov;23(11):715-720.