Cystic fibrosis (CF) is a rare autosomal recessive disorder caused by mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) gene located on the long arm of human chromosome 7,1,2 The CFTR gene encodes a protein in the cell membranes of the lungs, liver, pancreas, skin sweat glands, and reproductive organs.3,4 The CFTR protein is essential for the transportation of chloride ions and water molecules. Mutations of the CFTR gene cause CFTR protein dysfunction, resulting in decreased secretion of chloride and increased reabsorption of sodium and water across the epithelial cells. The consequent accumulation of thickened, purulent, and dehydrated secretions leads to airway obstruction, recurrent bacterial infections, and chronic airway inflammation.3,5–7
The organs and systems most affected by CF include the respiratory tract, pancreas, gastrointestinal tract, sweat glands, and reproductive system. This progressive and often lethal disease affects approximately 1 in 25 000 newborns in the Caucasian populations, with over 2000 CFTR mutations described.8,9 Of these, the p.Phe508del mutation accounts for 70–90% of CF cases in Caucasians.10 Data regarding non-Caucasians is limited, particularly for Arab and Asian populations. CF has been under-recognized in such groups due to lower awareness of this disease among pediatricians, its relative rarity, limited diagnostic resources, paucity of physicians specialized in CF care, and the lack of appropriate drugs.11
The prevalence of CF in the Omani population is still unknown. Based on retrospective and prospective analyses of cases in the North Al-Batinah governorate between 1998 and 2012, Fass et al,12 predicted the prevalence in Oman to be 1 in 8264; they also estimated the carrier frequency in the North Al-Batinah region to be 3.9 times higher than that in the total Omani population (1 in 29 vs. 1 in 94). However, because the study was confined to a single region, they could not precisely determine the national prevalence of CF.11 A mutational panel published in 2014 revealed that the commonest CFTR mutations among Omani children with clinical features of CF were p.Ser549Arg (S459R) and p.Phe508del (F508 del) (65.2% and 13%, respectively); other CF-causing mutations included c.2988+1G>A (3120+1G>A) (8.7%), p.Leu578Argfs* (4.3%), p.Ala357Thr (2.2%), and c.3718-2477C>T (3849+10kbC->T) (2.2%).13
The current study sought to narrow the data gap by describing the demographic distribution of CF in Oman, estimating its national prevalence, and providing updated mutational panels.
Methods
This retrospective cross-sectional study included all CF patients in Oman who were diagnosed and followed up between January 2006 and December 2020 at the two main tertiary hospitals in Oman, the Royal Hospital (RH) and Sultan Qaboos University Hospital (SQUH). These are the only centers in the country that diagnose and treat patients with CF. The diagnosis was based on the patient’s clinical presentation, high-sweat chloride test result, and the presence of two copies of CFTR mutations.14
We collected the required data from the electronic hospital databases of RH and SQUH. A predesigned Microsoft Excel spreadsheet was used to enter the demographic and clinical information for each patient, including date and place of birth, region, exact CFTR mutations and classes, family history of CF, parental consanguinity, and current health status. In cases with missing data, phone interviews were conducted with the patients or their parents or by accessing patient case notes in the respective regional hospital after obtaining verbal consent. The CFTR mutations and classes were classified according to the CFTR2 database.15
The data were statistically analyzed using IBM SPSS (IBM Corp. Released 2011. IBM SPSS Statistics for Windows, Version 20.0. Armonk, NY: IBM Corp.). Descriptive analyses of frequencies, percentages, averages, and SDs were performed. Statistical inferences were derived from two-tailed trials and the level of significance was set at p = 0.050. Cases were divided into three groups according to the year of birth as follows: before 2000, 2000–2009, and 2010–2020. Fisher’s exact test and Chi-squared test were used to compare the numbers of living and deceased cases over each decade. Prevalence was calculated based on the total population of Omani nationals < 30 years of age. This study received approval from the institutional research and ethics committees of both RH and SQUH.
Results
Between January 2006 and December 2020, a total of 227 CF patients were diagnosed and followed up at RH and SQUH. Of these, 53.7% were male and 46.3% were female. Positive sweat test findings were reported in 94.0% of the cohort, and CFTR mutations were identified in 86.3%. Parental consanguinity was observed in 68.3% while 69.6% had a family history of CF. The majority of the patients were from the Al-Batinah (52.0%), followed by the A’Dhahirah (19.8%), A’Dakhiliyah (11.5%), Muscat (6.6%), A’Sharqiyah (5.7%), and Dhofar (4.4%) [Table 1].
Table 1: Characteristics of confirmed cystic fibrosis patients (N = 227).
|
Sex
|
|
|
|
|
|
|
|
|
Male
|
16
|
15
|
64
|
2
|
4
|
21
|
122 (53.7)
|
|
Female
|
11
|
14
|
59
|
2
|
3
|
16
|
105 (46.3)
|
|
Status
|
|
|
|
|
|
|
|
|
Alive
|
18
|
22
|
108
|
2
|
7
|
9
|
166 (73.1)
|
|
Died
|
8
|
7
|
15
|
0
|
0
|
23
|
53 (23.3)
|
|
Unknown
|
1
|
0
|
0
|
2
|
0
|
5
|
8 (3.5)
|
|
Family history
|
|
|
|
|
|
|
|
|
Yes
|
20
|
22
|
91
|
1
|
4
|
20
|
158 (69.6)
|
|
No
|
6
|
6
|
31
|
1
|
3
|
12
|
59 (26.0)
|
|
Consanguinity
|
|
|
|
|
|
|
|
|
Yes
|
24
|
23
|
93
|
1
|
0
|
14
|
155 (68.3)
|
|
No
|
3
|
4
|
13
|
0
|
7
|
3
|
30 (13.2)
|
|
Unknown
|
0
|
2
|
17
|
3
|
0
|
20
|
42 (18.5)
|
|
Region
|
|
|
|
|
|
|
|
|
Al-Batinah
|
13
|
22
|
67
|
0
|
2
|
14
|
118 (52.0)
|
|
A’Dhahirah
|
1
|
1
|
37
|
0
|
0
|
6
|
45 (19.8)
|
|
A’Dakhiliyah
|
7
|
0
|
14
|
0
|
3
|
2
|
26 (11.5)
|
|
Muscat
|
3
|
4
|
1
|
0
|
0
|
7
|
15 (6.6)
|
|
A’Sharqiyah
|
3
|
2
|
4
|
1
|
0
|
3
|
13 (5.7)
|
*CFTR classes: class I: protein synthesis defect; class II: maturation defect; class III: gating defect; class IV: conductance defect; class V: reduced quantity;
class VI: reduced stability.
At the end of our study period, December 2020, 166 (73.1%) patients were alive, 53 (23.3%) died, and eight (3.5%) were lost to follow-up. Of the CF patients born prior to 2000, 47.7% were still a live, compared to 70.2% of those born between 2000–2010, and 90.8% of those born between 2010–2020 (p < 0.001) [Figure 1]. Based on our data the prevalence of CF in Oman is 10.3 per 100 000 individuals.

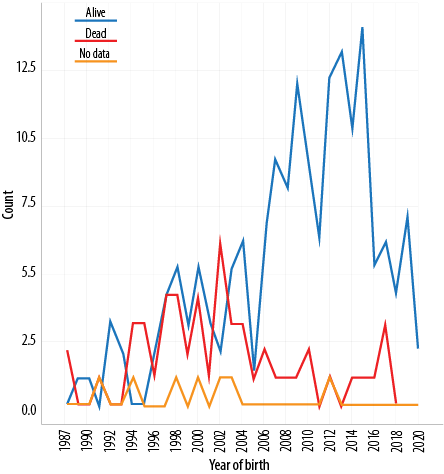 Figure 1: Distribution of cases according to the year of birth and outcome status (N = 227).
Figure 1: Distribution of cases according to the year of birth and outcome status (N = 227).
The most common CFTR mutation was p.Ser549Arg (both homozygous and heterozygous), followed by p.Phe508del (both homozygous and heterozygous) and c.2988+1G>A (both homozygous and heterozygous), affecting 51.9%, 12.3%, and 4.4% of the cohort, respectively. Other mutations accounted for 24.4% of cases while 7.0% had no genetic tests done at the time of the study [Table 2]. In particular, Leu88TyrFs* (c.263delT), p.Asp192Val (c.575A>T), and c.4242+1G>C were identified as novel CFTR mutations in our study. In addition, p.Leu578Argfs* (c.1733-1734delTA) and p.Ala357Thr (c.1069G>A) mutations reported in a previous study as novel mutations, were also identified in our cohort.13
Table 2: Different cystic fibrosis transmembrane conductance regulator (CFTR) mutation classes and their prevalence among Omani children with confirmed genetic diagnoses of cystic fibrosis (N = 227).
|
1
|
S549R/ homo
|
c.1647T>G
|
p.Ser549Arg
|
III
|
118
|
52.0
|
|
2
|
F508del / homo
|
c.1521_1523delCTT,
|
p.Phe508del
|
II
|
28
|
12.3
|
|
3
|
3120+1G->A
|
c.2988+1G>A
|
-
|
I
|
10
|
4.4
|
|
4
|
A357T
|
c.1069G>A
|
p.Ala357Thr
|
Novel/unknown
|
2
|
0.9
|
|
5
|
L578delTA
|
c.1733-1734delTA
|
p.Leu578Argfs*
|
I
|
3
|
1.3
|
|
6
|
K1177X homo
|
c.3529A>T
|
p.Lys1177X
|
I
|
4
|
1.8
|
|
7
|
V392G / S549R
|
c.1175T>G / c.1647T>G
|
p.Val392Gly / p.Ser549Arg
|
III
|
4
|
1.8
|
|
8
|
S549R / F508del
|
c.1647T>G / c.1521_1523delCTT
|
p.Ser549Arg / p.Phe508del
|
III/II
|
1
|
0.4
|
|
9
|
F508del / 3120+1G->A
|
c.1521_1523delCTT / c.2988+1G>A
|
p.Phe508del / –
|
II/I
|
2
|
0.9
|
|
10
|
1525-1G>A / R560S
|
c.1393-1G>A / c.1680A>C
|
– / p.Arg560Ser
|
I/Unknown
|
1
|
0.4
|
|
11
|
cystic fibrosis transmembrane conductance regulator
|
c.850dupA
|
p.Met284Asnfs*
|
I
|
3
|
1.3
|
|
12
|
cystic fibrosis transmembrane conductance regulator
|
c.263delT
|
Leu88TyrFs*
|
I
|
4
|
1.8
|
|
13
|
W361R / –
/ F1052V
|
c.11801T>C / c.3154T>G / c.263delT
|
p.Trp361Arg / p.Phe1052Va l / Leu88TyrFs*
|
II/IV/I
|
3
|
1.3
|
|
14
|
3849+10kbC>T
|
c.3718-2477C>T
|
-
|
IV
|
4
|
1.8
|
|
15
|
D192V
|
c.575A>T
|
p.Asp192Val
|
Unknown
|
2
|
0.9
|
|
16
|
4374+ 1G->T
|
C.4242+1G>C
|
-
|
I
|
2
|
0.9
|
|
17
|
CFTRdele4-11
|
c.(273+1_274-1)_ (1679+1_1680-1)del
|
-
|
I
|
1
|
0.4
|
|
18
|
2118del4 /S945L
|
c.1986_1989delAACT/ c.2834C>T
|
p.Thr663Argfs*/ p.Ser945Leu
|
I/IV
|
2
|
0.9
|
|
19
|
K1177X / D192V
|
c.3529A>T / c.575A>T
|
p.Lys1177X /
p.Asp192Val
|
I/Unknown
|
1
|
0.4
|
|
20
|
S549R / S945L
|
c.1647T>G / c.2834C>T
|
p.Ser549Arg / p.Ser945Leu
|
III
|
1
|
0.4
|
Discussion
There is limited research regarding the prevalence and nature of CF in Arab populations. As in the other Middle Eastern countries, CF has historically been underdiagnosed and underestimated in Oman.11 With recent advances in molecular genetic testing and sequencing and growing awareness of CF among health providers, additional novel CF-causing mutations have been identified. With new biochemical and genetic diagnostic facilities available, significantly more CF cases have been detected in Oman in the last two decades.
The current study aimed to describe the demographic distribution of CF in Oman, estimate the national prevalence of the disease, and provide updated mutational panels. Because this study included the only two centers in the country to provide care for patients with CF, we believe that the estimated prevalence rate in this study is nationally representative. However, our calculation of the incidence rate of CF may not be accurate as the number of newly diagnosed cases each year has varied between 3 and 10. In addition, several patients were lost to follow-up and their outcomes could not be ascertained.
Previous studies conducted in other Arab countries have given a widely differing prevalence of CF. While in Kuwait it was estimated to be 0.7–1.0 per 100 000 population, in Bahrain it was 3 per 100 000.16–18 A Saudi Arabian study reported a higher prevalence at 23.7 per 100 000.18,19 UAE reported an estimated prevalence of 6.3 per 100 000.20,21 Comparatively, the prevalence of CF in Oman, at 10.3 per 100 000, seems moderate. One of the factors responsible for such wide variations in reported CF prevalence between Arabian Gulf countries could be the lack of national CF registries. In addition, these studies are several decades old and may not reflect the current prevalence rates; as the recorded numbers of rare diseases like CF have been increasing over time due to advances in diagnostic definitions, technologies, treatments, awareness, and expertise.19
The findings of the mutational panel of 2014 by Al Kindy et al,13 in the Omani population indicated a wide and variable heterogeneity in CFTR mutations, totaling six mutations. With new findings in the present study, the mutational panel now features 20 CFTR mutations. This spectrum of mutations in the Omani population overlaps with those found in other Arab and European populations.12,13,20–23 The commonest CFTR mutation in our study was p.Ser549Arg (a class III gating mutation), common in the Gulf Cooperation Council region.12,13 This mutation has been reported in CF patients of Arab Bedouin and Moroccan descent.23 Interestingly, p.Ser549Arg is rare in other ethnicities, accounting for only 1% of their CF cases.8,20
In the UAE population, p.Ser549Arg and p.Phe508del are reported to be the commonest CFTR mutations.20–22 While p.Phe508del accounts for 70–90% of CFTR mutations in Western populations, it was prevalent only in 12.3% of the Omani children we studied, making it the second commonest mutation in this country.8,10
In 2000, Frossard et al,22 found that Omani patients of Bedouin descent carried the p.Ser549Arg mutation, whereas those of Baluch descent had the p.Phe508del mutation (homozygous). This finding was supported by earlier research.13 The p.Phe508del mutation has also been described in 56% of Tunisian and Northwest African CF patients.24 The same mutation has also been identified in 34% of Lebanese CF patients, but only in 8% of CF patients in Oman’s neighbor, Bahrain.23
We found c.2988+1G>A to be the third most common mutation in Omani CF patients, supporting the earlier report by Al Kindy et al.13 Omani patients are unique here because c.2988+1G>A is much rarer in other Middle Eastern countries such as Jordan, Saudi Arabia, Qatar, and the UAE.23 Curiously, this mutation is well described and reported with high frequency in African populations. This shared mutation among Omani and African CF patients is possibly a legacy of Oman’s history as a maritime empire with territories in East Africa, which may have caused centuries of ethnic mixing.13,23
Using the modern genetic testing facilities in Oman, we sequenced the entire CFTR gene and identified several novel mutations. These included Leu88TyrFs*, p.Asp192Val, and c.4242+1G>C, alongside two mutations previously identified by Al-Kindy et al,13 (p.Leu578Argfs* and p.Ala357Thr).
The geographical distribution of CF in Oman is not uniform. Most of our CF patients were from Al-Batinah governorate, followed by the A’Dhahirah and A’Dakhiliyah governorates. Moreover, 64.3% of CF patients carrying p.Ser549Arg and p.Phe508del CFTR mutations—the two commonest mutations—were clustered in Al-Batinah and A’Dhahirah, while other less frequent mutations were clustered in other governorates. Furthermore, we have observed A’Dakhiliyah CF patients carrying the rest of heterogeneous CFTR mutations. Class IV mutation c.3718-2477C>T (3849+10kbC>T) was found solely in CF patients from Dhofar, the southernmost governorate of Oman, all of whom had normal sweat chloride levels. Interestingly, three CF- affected children in a single family had three CFTR mutations present as a compound heterozygous (p.Trp361Arg / p.Phe1052Val / Leu88TyrFs*). The segregation study of both parents showed that the mother, though asymptomatic, was a carrier of two variants p.Trp361Arg and p.Phe1052Val located in a single allele as cis. The father was a carrier for Leu88TyrFs*. Such prevalence of clustered CF-causing alleles found in specific communities in Oman may also be explained by the region’s history of frequent immigration and passage from East Africa, the Indian subcontinent, and beyond.
The tendency for consanguinity, deeply rooted in the Arabian culture, is a magnifying factor for genetic diseases. The consanguineous marriage rate in our cohort was 68.3%. Most patients (69.6%) had one or more family members or relatives diagnosed with CF. Oman has a unique population structure in which many rural tribal communities remain established in their traditional places of residence, increasing the likelihood of marriages between blood relations. As a result, Omanis have a relatively higher frequency of genetic disorders.25–28
We did not identify any CF patients originating from Musandam, a governorate of Oman that is non-contiguous with the mainland, but close to the UAE. It is likely that Omani CF patients from Musandam access health services in the UAE.
Considering the high prevalence of CF in Oman, exacerbated by the practice of consanguinity, we recommend a newborn screening program facilitate early diagnosis of genetic disorders such as CF to improve the odds of survival and wellbeing of the affected children. In areas with higher prevalence rates of CF, healthcare providers should be instructed to inculcate the importance of premarital genetic screening and to recommend preimplantation genetic diagnosis before in-vitro fertilization.
Limitations of this study included the difficulty to calculate the incidence of CF due to the lack of a national CF registry, the lack of newborn screening program, missing or partial data, and the absence of accurate methods of detection to enable early diagnosis. The prevalence of CF reported in this study could thus be an underestimation.
Conclusion
The estimated prevalence of CF in Oman is 10.3 per 100 000 individuals. A total of 21 disease-causing CFTR mutations have been identified, of which p.Ser549Arg and p.Phe508del were the most common. Three novel mutations have also been detected. In terms of geographical distribution, CF cases were clustered mainly in Al-Batinah and A’Dhahirah governorates. Premarital genetic counseling and preimplantation genetic testing are highly recommended in CF-prevalent governorates of Oman.
Disclosure
The authors declared no conflicts of interest. No funding was received for this study.
references
- 1. Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989 Sep;245(4922):1066-1073.
- 2. Rubenstein RC. Targeted therapy for cystic fibrosis: cystic fibrosis transmembrane conductance regulator mutation-specific pharmacologic strategies. Mol Diagn Ther 2006;10(5):293-301.
- 3. Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989 Sep;245(4922):1073-1080.
- 4. Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005 May;352(19):1992-2001.
- 5. Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med 2007;58:157-170.
- 6. Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, et al; The Pulmozyme Study Group. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. N Engl J Med 1994 Sep;331(10):637-642.
- 7. Quan JM, Tiddens HA, Sy JP, McKenzie SG, Montgomery MD, Robinson PJ, et al; Pulmozyme early intervention trial study group. A two-year randomized, placebo-controlled trial of dornase alfa in young patients with cystic fibrosis with mild lung function abnormalities. J Pediatr 2001 Dec;139(6):813-820.
- 8. Hospital for Sick Children. Cystic fibrosis mutation database. 2011 [cited 2021 October 12]. Available from: www.genet.sickkids.on.ca/AdvancedSearchPage.html.
- 9. Cystic Fibrosis Foundation. Know your mutations: a CFTR mutation fact sheet. 2017 [cited 2021 October 12]. Available from: https://cff.org/What-is-CF/Genetics/Know-Your-CFTR-Mutations-Infographic.pdf.
- 10. De Boeck K, Zolin A, Cuppens H, Olesen HV, Viviani L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J Cyst Fibros 2014 Jul;13(4):403-409.
- 11. Kabra SK, Kabra M, Shastri S, Lodha R. Diagnosing and managing cystic fibrosis in the developing world. Paediatr Respir Rev 2006;7(Suppl 1):S147-S150.
- 12. Fass UW, Al-Salmani M, Bendahhou S, Shivalingam G, Norrish C, Hebal K, et al. Defining a mutational panel and predicting the prevalence of cystic fibrosis in oman. Sultan Qaboos Univ Med J 2014 Aug;14(3):e323-e329.
- 13. Al-Kindy H, Ouhtit A, Al-Salmi Q, Al-Bimani M, Al-Nabhani M, Gupta I. Novel mutation in the CFTR gene of cystic fibrosis patients in Oman. J Mol Biomark Diagn 2014;5(2):1000168.
- 14. Rosenstein BJ, Cutting GR; Cystic Fibrosis Foundation Consensus Panel. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998 Apr;132(4):589-595.
- 15. Cystic Fibrosis Foundation, Johns Hopkins University, Hospital for Sick Children. The clinical and functional translation of CFTR (CFTR2) database. 2011 [cited 2021 October 12]. Available from: http://cftr2.org.
- 16. Al-Mahroos F. Cystic fibrosis in bahrain incidence, phenotype, and outcome. J Trop Pediatr 1998 Feb;44(1):35-39.
- 17. Kollberg H. Cystic fibrosis in Kuwait. J Trop Pediatr 1986 Dec;32(6):293-294.
- 18. Hammoudeh S, Gadelhaq W, Hani Y, Omar N, El Dimassi D, Elizabeth C, et al. The epidemiology of cystic fibrosis in Arab countries: a systematic review. SN Compr Clin Med 2021;3(2):490-498.
- 19. Banjar H. Mortality data for cystic fibrosis patients in a tertiary care center in Saudi Arabia. Ann Saudi Med 2003 Nov-Dec;23(6):416-417.
- 20. Dawson KP, Frossard PM. Cystic fibrosis in the United Arab Emirates: an under-recognized condition? Trop Doct 1995 Jul;25(3):110-111.
- 21. Frossard PM, Lestringant G, Girodon E, Goossens M, Dawson KP. Determination of the prevalence of cystic fibrosis in the United Arab Emirates by genetic carrier screening. Clin Genet 1999 Jun;55(6):496-497.
- 22. Frossard PM, Dawson KP, Das SJ, Alexander PC, Girodon E, Goossens M. Identification of cystic fibrosis mutations in Oman. Clin Genet 2000 Mar;57(3):235-236.
- 23. Center of Arab Genomic Studies. Catalogue for Transmission Genetics in Arabs (CTGA) database. 2009 [cited 2021 October 12]. Available from: https://cags.org.ae/en/search-database#SearchAdvanced.
- 24. Boussetta K, Khalsi F, Bahri Y, Belhadj I, Tinsa F, Messaoud TB, et al. Cystic fibrosis in Tunisian children: a review of 32 children. Afr Health Sci 2018 Sep;18(3):664-670.
- 25. Rajab A, Bappal B, Al-Shaikh H, Al-Khusaibi S, Mohammed AJ. Common autosomal recessive diseases in Oman derived from a hospital-based registry. Community Genet 2005;8(1):27-30.
- 26. Rajab A, Al Rashdi I, Al Salmi Q. Genetic services and testing in the Sultanate of Oman. Sultanate of Oman steps into modern genetics. J Community Genet 2013 Jul;4(3):391-397.
- 27. Rajab A, Al Salmi Q, Jaffer J, Mohammed AJ, Patton MA. Congenital and genetic disorders in the Sultanate of Oman. First attempt to assess healthcare needs. J Community Genet 2014 Jul;5(3):283-289.
- 28. Rajab A, Patton MA. A study of consanguinity in the Sultanate of Oman. Ann Hum Biol 2000 May-Jun;27(3):321-326.