Multi-system involvement is the hallmark of syndrome described and named after Gordon Sharp and his colleagues.1 It is an inflammatory disease characterized by abnormal B- and T-cell response to apoptotic ally modified nuclear antigens in the messenger RNA, most commonly the U1-ribonucleoprotein (RNP). It typically affects women between the ages of 15 and 30 years. Not only is neurological involvement rare in autoimmune disorders, it is exceptionally rare in mixed connective tissue disease (MCTD). The manifestations are variable, making the diagnosis and management challenging. These challenges are highlighted in our case report, which presented with mixed polyneuropathy.
Case report
A 36-years-old Omani gentleman presented to the Emergency Department at the Sultan Qaboos University Hospital with a six-month history of gradual lower leg weakness and swelling with the inability to walk. In the preceding year, he was hospitalized on multiple occasions, both locally and abroad, for polyarthralgia, low back pain, and paresthesia involving both feet. His other comorbidities included difficult to control hypertension, for which he was on three separate classes of anti-hypertensive medications. The patient was admitted previously with hypertensive encephalopathy requiring high dependency care, from which he made an uneventful recovery. In addition, he had lumbar spine degenerative disc disease. There was no significant family history of connective tissue disorders. He was unable to continue his job in construction due to his symptoms. He was also a heavy smoker and previously consumed alcohol of undisclosed duration and quantity.
On examination in the emergency department, he looked unwell, with a Glasgow Coma Scale score of 15/15. He was afebrile with a regular pulse of 82 beats per minute, a transcutaneous O2 saturation of 98% on room air, and blood pressure of 140/88 mmHg. His peripheries were cold but well perfused with no Raynaud’s related changes. He had no joint synovitis, skin rash, or mucocutaneous ulcers. His cardio-respiratory examination was unremarkable apart from loud pulmonic component of the second heart sound on auscultation. The neurological examination was remarkable for bilateral distal muscle weakness in a symmetrical distribution with severe bilateral foot drop. There was absent ankle dorsiflexion, plantarflexion, inversion, and eversion. Extension and flexion at the knee joint were estimated at 3/5 power. More proximally, he could perform hip flexion, extension, abduction, and adduction at the hip joint with grade 4/5 power. Furthermore, when testing pinprick sensation and temperature, both modalities were absent bilaterally in a symmetrical distribution extending up the knees. These modalities were also tested and noted to be symmetrically diminished to a milder extent in both hands (gloves-and-stockings distribution of sensory deficits). He had marked lower extremity pitting edema extending up to his knees. Bedside urine dipstick was positive 2+ for protein.
His blood investigations during the hospital stay are summarized in Table 1. The radiological workup included a brain magnetic resonance imaging, which showed diffuse brain atrophy but no focal pathology, and a computed tomography chest scan revealing areas of septal and centrilobular emphysema of upper lobes with prominent pulmonary trunk and both main pulmonary arteries. His transthoracic echocardiography demonstrated moderate concentric left ventricle hypertrophy with normal systolic function and severe pulmonary hypertension (estimated pulmonary artery systolic pressure of 62 mmHg). A nerve condition study revealed absent motor and sensory signals in both lower limbs and a mild reduction of sensory amplitude in upper limbs. Electromyography of sampled muscles in lower extremities showed denervation and neurogenic action potentials. In summary, the physiological data was suggestive of sensori-motor axonal neuropathy affecting predominantly the lower extremities.
Table 1: Summary of pertinent lab investigations.
|
Hemoglobin |
10.5 |
11–14.5 gm/dL |
|
Erythrocyte sedimentation rate |
115 |
0–30 mmL/hr |
|
Direct antiglobulin test (Coombs Test) |
Positive |
|
|
C-reactive protein |
24 |
0–5 mg/L |
|
Creatinine |
108 |
45–84 umol/L |
|
Complement C3 |
0.94 |
0.90–1.80 g/L |
|
Complement C4 |
0.27 |
0.10–0.40 g/L |
|
Red blood cell folate |
25.9 |
7–46.4 nmol/L |
|
Vitamin B12 |
1476 |
138–652 pmol/L |
|
Creatine kinase |
31 |
39–308 U/L |
|
Anti-nuclear antibody |
1:640 speckled |
|
|
Extractable nuclear antigens |
Strongly positive anti-RNP |
|
|
Myositis screening |
Negative |
|
|
Anti-ds DNA antibodies |
Negative |
|
|
Anti-phospholipid antibodies |
Negative |
|
Given the strongly positive antinuclear antibody and anti-U1-RNP, high inflammatory markers, and multisystemic involvement, the patient was diagnosed with MTCD and was pulsed with IV methylprednisolone. A kidney biopsy was suggested for further evaluation given nephrotic range proteinuria. Unfortunately, he refused a kidney biopsy but consented to a sural nerve biopsy. Careful evaluation of the histopathological slides revealed extensive degenerative changes, including myelin-ovoid formation and axonal fragmentation [Figure 1]. More importantly, prominent peri-vascular and transmural lymphocytic inflammation and occlusion were consistent with vasculitis [Figure 2].

Figure 1: Histopathological section from the sural nerve biopsy specimen showing extensive degenerative changes, including myelin-ovoid formation and axonal fragmentation.
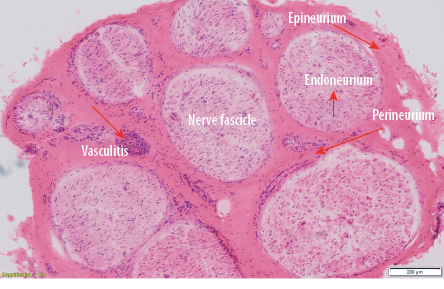
Figure 2: Histopathological section from the sural nerve biopsy specimen showing perivascular and transmural lymphocytic inflammatory infiltrate and vessel occlusion, consistent with vasculitis magnification = × 10.
After pulse therapy, he was placed on a tapering course of prednisolone in addition to steroid sparking agents, including hydroxychloroquine and mycophenolate mofetil. He also received nutritional support and physiotherapy in foot drop splints with tone and power restoring exercises. Right heart catheterization revealed moderate pulmonary hypertension (mean pulmonary artery pressure of 38 mmHg and mean pulmonary capillary wedge pressure of 7 mmHg) with negative vasoreactivity and was treated with sildenafil.
Discussion
The neurological involvement tends to be mild in patients with MCTD.2 Trigeminal neuralgia is the most common neurological manifestation,3 but there are published reports of more severe presentations such as central nervous system vasculitis4 or aseptic meningitis.2 The pathogenesis of polyneuropathy remains unclear, but it is likely related to peripheral neuritis with regional vasculitis, as shown on nerve biopsy.5 There is no standard regimen to manage MCTD related polyneuropathy.6 Therapy follows standard guidelines on managing systemic lupus erythematosus and other connective
tissue diseases.6,7
At first glance, the biopsy showed widespread degenerative changes, which would have been consistent with a severe nutritional deficiency, especially in a chronic alcoholic. However, our patient had normal hemoglobin, red blood cell folate, and vitamin B12 levels with no evidence of megaloblastosis, which made nutritional deficiency less likely to cause severe sensory-motor deficits.
On more careful evaluation, there was clear evidence of vasculitis with vessel wall inflammation, inflammatory cell infiltrates, and occlusion. This highlights the importance of the meticulous evaluation of biopsy specimens within the clinical context. In addition, it is conceivable that such difficult-to-spot changes may be missed on an inadequate biopsy specimen.
The natural history of MCTD related polyneuropathy is unclear. However, it appears our patient made a near-complete recovery with immunosuppressive therapy. Further studies on the outcome of such patients are needed but likely to be quite challenging given the rarity of the disease.
Conclusion
Despite being unusual, severe polyneuropathy could be an initial neurological presentation of MCTD. The diagnosis can be challenging and requires extensive and careful workup, including neurophysiology and nerve biopsy. In addition, immunosuppressive therapy and extensive rehabilitation are integral components
of management.
Disclosure:
The authors declared no conflicts of interest. Written consent was obtained from the patient.
references
- 1. Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR. Mixed connective tissue disease–an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med 1972 Feb;52(2):148-159.
- 2. Nowicka-Sauer K, Czuszynska Z, Majkowicz M, Smoleńska Ż, Jarmoszewicz K, Olesińska M, et al. Neuropsychological assessment in mixed connective tissue disease: comparison with systemic lupus erythematosus. Lupus 2012;21:927-933.
- 3. Nimelstein SH, Brody S, McShane D, Holman HR. Mixed connective tissue disease: a subsequent evaluation of the original 25 patients. Medicine (Baltimore) 1980;59:239-248.
- 4. Narváez J, Pascual M, Albert Espi G, Millan M, López de Recalde M, Alegre JJ, Castellví I, Gomez Vaquero C, Nolla JM. Prevalence of Organ Involvement in Mixed Connective Tissue Disease. Arthritis Rheumatol 2017;69 (suppl 10).
- 5. Hui M, Meena AK, Rajasekhar L, Sireesha Y, Afshan J, Mridula R, et al. Vasculitic Neuropathy: A Retrospective Analysis of Nerve Biopsies and Clinical Features from a Single Tertiary Care Center. Ann Indian Acad Neurol 2019;22(2):180-186.
- 6. Gunnarsson R, Hetlevik SO, Lilleby V, Molberg O. Mixed connective tissue disease. Best Practice & Research Clinical Rheumatology 2016;30(1):95–111.
- 7. Ciang NC, Pereira N, Isenberg DA. Mixed connective tissue disease-enigma variations? Rheumatology (Oxford) 2017 Mar;56(3):326-333.