Heterozygous β-thalassemia patients generally are asymptomatic and non-anemic compared to homozygous β-thalassemia patients who most of the time presented with a severe form of anemia and worse clinical presentations.1 The molecular defects of β-thalassemia results in absent or reduced β-chain production while alpha chain (α-chain) synthesis is unaffected. As in homozygous β-thalassemia, excess α-chain leads to its precipitation in red cell precursors, forming an intracellular inclusion that leads to ineffective erythropoiesis and hemolysis. However, in heterozygous β-thalassemia, only minimal excess of α-globin chain was produced, resulting in mild anemia without much clinical significance.2
β-thalassemia exhibits remarkable phenotypic variability, ranging from mild condition to severe anemia that requires regular blood transfusions. The coinheritance of α-thalassemia results in milder clinical manifestations due to the reduction in α-globin chain excess.3 In contrast, the additional α-globin chain in β-thalassemia heterozygous increases the chain imbalance, converting a typically asymptomatic state of heterozygous β-thalassemia to thalassemia intermedia.4
We report a pregnant woman with thalassemia intermedia phenotype due to coinheritance of heterozygous β-globin chain mutation (IVS1-5) and a very rare heterozygous α-globin triplication (ααα anti-3.7), which could not be identified by standard hemoglobin (Hb) analysis. This case focuses on its clinico-hematological features and diagnostic approach, emphasizing the role of molecular testing in making the diagnosis.
Case report
A 32-year-old Malay woman, gravida 3 para 2 at 25-week pregnancy presented with jaundice. She had a history of multiple episodes of jaundice since secondary school and during her two previous pregnancies, but it was never investigated further. She required four to six pints of red cells transfusion during each pregnancy.
Clinically, she was jaundiced and pale with no features of thalassemic face. There was no hepatosplenomegaly. The initial blood investigation results were: red blood cell (RBC) count of 3.01 × 1012/L, Hb of 7.2 g/dL with a mean corpuscular volume (MVC) of 74.2 fL, mean corpuscular Hb (MCH) of 23.3 pg, and a red cell distribution width (RDW) of 22.5%. Peripheral blood smear showed hypochromic microcytic RBCs with anisopoikilocytosis, basophilic stippling, presence of normoblasts, and polychromatic cells with reticulocytes count of 6%. Her serum ferritin was 190 μg/L. Other investigations showed lactate dehydrogenase (LDH) of 287 U/L, total bilirubin of 111.7 umol/L, and normal liver enzymes. Coombs test was negative. Viral screening for hepatitis B and C was negative.
The result of Hb analysis using SEBIA Hb Capillary Electrophoresis was consistent with β-thalassemia trait: HbA 91.2%, HbA2 5.1%, and HbF 3.7%. Given her clinical presentation of chronic hemolysis and recurrent transfusions (thalassemia intermedia), further molecular studies; multiplex gap-polymerase chain reaction (PCR) for α-gene deletion (-α3.7, -α4.2, --SEA, --MED, -(α)20.5, and --THAI), multiplex Amplification Refractory Mutation System RMPCR for β-gene mutation, and multiplex PCR for α-globin gene mutation were done.5 The molecular results of β -globin gene revealed heterozygous IVS1-5 (β+) mutation. The result of DNA analysis for α-globin gene/cluster identified was a triple alpha-globin chain (αααanti-3.7) mutation [Figure 1]. There was no deletional α-thalassemia detected.
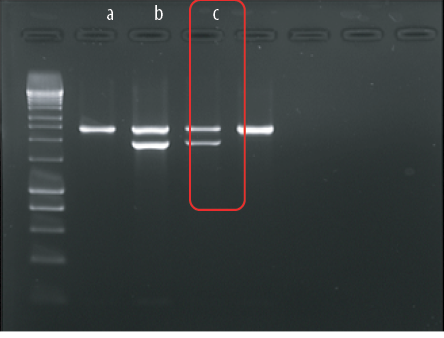
Figure 1: Alpha triplication multiplex polymerase chain reaction of (a) negative control, (b) positive control αααanti-3.7, and (c) the index patient. Presence of αααanti-3.7 in the patient sample as in positive control.
Discussion
The genetic testing of the patient revealed the combination of triplication of α-globin gene with heterozygous β-thalassemia had produced a thalassemia intermedia phenotype, as it increases the imbalance between the α- and β-globin chains.
The deletional types of α-thalassemia are much more common worldwide compared to α-globin gene triplication.6 The coexistence of α-gene triplication is an important modifier of the severity of β-thalassemia trait or β-thalassemia intermedia. It may exacerbate the phenotypic severity of β-thalassemia by causing severe anemia with significant clinical manifestation by causing more globin chain imbalance.7
These genetic and clinical findings have important implications for prenatal screening and genetic counseling programs. The coinheritance of extra α-globin chain and β-thalassemia carriers are at risk of having an affected offspring, although the partners may be entirely normal. Thus, detecting the α-globin gene triplication with β-thalassemia would help physicians provide appropriate genetic counseling and a proper management plan. This will assist physicians in illustrating the possible implications of the disease to the patients.8
This case is highlighted due to relatively rare α-gene triplication compared to α-gene deletion and its coinheritance with β-thalassemia trait that worsens the clinical and hematological features of the patient.
Conclusion
The information given in this case report could be used as guidance for the diagnosis and management of heterozygous β-thalassemia in patients with the intermedia phenotype.
Disclosure
The authors declared no conflicts of interest. Written informed consent was obtained from the patient and her kin.
references
- 1. Galanello R, Ruggeri R, Paglietti E, Addis M, Melis MA, Cao A. A family with segregating triplicated alpha globin loci and beta thalassemia. Blood 1983 Nov;62(5):1035-1040.
- 2. Traeger-Synodinos J, Kanavakis E, Vrettou C, Maragoudaki E, Michael T, Metaxotou-Mavromati A, et al. The triplicated alpha-globin gene locus in beta-thalassaemia heterozygotes: clinical, haematological, biosynthetic and molecular studies. Br J Haematol 1996 Dec;95(3):467-471.
- 3. Farashi S, Bayat N, Faramarzi Garous N, Ashki M, Montajabi Niat M, Vakili S, et al. Interaction of an α-Globin gene triplication with β-Globin gene mutations in iranian patients with β-Thalassemia intermedia. Hemoglobin 2015;39(3):201-206.
- 4. Camaschella C, Kattamis AC, Petroni D, Roetto A, Sivera P, Sbaiz L, et al. Different hematological phenotypes caused by the interaction of triplicated alpha-globin genes and heterozygous beta-thalassemia. Am J Hematol 1997 Jun;55(2):83-88.
- 5. Wang W, Ma ES, Chan AY, Prior J, Erber WN, Chan LC, et al. Single-tube multiplex-PCR screen for anti-3.7 and anti-4.2 α-globin gene triplications. Clin Chem 2003 Oct;49(10):1679-1682.
- 6. Liu YT, Old JM, Miles K, Fisher CA, Weatherall DJ, Clegg JB. Rapid detection of alpha-thalassaemia deletions and alpha-globin gene triplication by multiplex polymerase chain reactions. Br J Haematol 2000 Feb;108(2):295-299.
- 7. Naderi M, Miri-Moghaddam I, Dorgalaleh A, Alizadeh S, Tabibian S, Pishjoo M. A patient with coinheritance of alpha-globin gene triplication and IVSI-5 mutation of beta-globin gene. Zahedan journal of research in medical sciences 2015 May 31;17(5):1-3.
- 8. Cebrian FY, Flores MD, Álvarez SI, Salinas IP, Iturrate CR. Combination of a triple alpha-globin gene with beta-thalassemia in a gypsy family: importance of the genetic testing in the diagnosis and search for a donor for bone marrow transplantation for one of their children. BMC Res Notes 2016 Apr;9:220.