Cochlear incomplete partition type III anomaly (IP-III), also referred to as X-linked deafness type 2 or X-linked stapes gusher syndrome, is a rare congenital cause of severe progressive mixed hearing loss affecting mainly males.1 It has characteristic radiological features, consisting of a corkscrew appearance of the cochlea due to dilatation of the internal auditory canal (IAC) and the direct communication between the IAC and the cochlea secondary to the absent bony plate between the two cavities (lamina cribrosa).2,3 Preoperative diagnosis of IP-III facilitates the surgeon in avoiding unnecessary stapes surgery and possible complications of cochlear implantation, such as perilymph gushing during cochleostomy and misplacement of the electrode into the internal auditory meatus, which can lead to further hearing loss. Furthermore, the preoperative computed tomography images facilitate the surgeon in sizing and selecting the most appropriate implant type for the procedure.2
Case report
A 10-month-old male infant presented for the first time with his parents expressing concerns about his poor response to sound stimuli by not turning towards soft or loud sound sources. The child’s past medical and surgical history was none contributory with an uneventful antenatal period followed by normal full-term delivery. As reported by the mother, he underwent an otoacoustic emissions test at the time of birth and passed. No family history of hearing loss was reported. Physical examination revealed profound sensorineural hearing loss bilaterally. The auditory brainstem response test showed profound hearing loss at 85 decibels above normal adult hearing level (dBnHL) on the left and 90 dBnHL on the right.
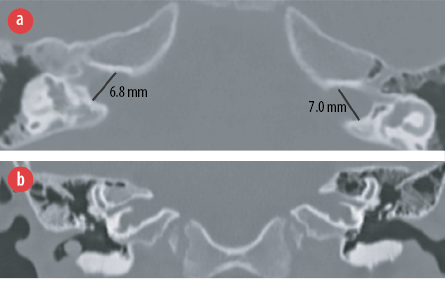
Figure 1: CT coronal section of the internal auditory meatus (IAM) demonstrating (a) bulbous dilation of the IAM and (b) corkscrew cochlea and absent lamina cribrosa.
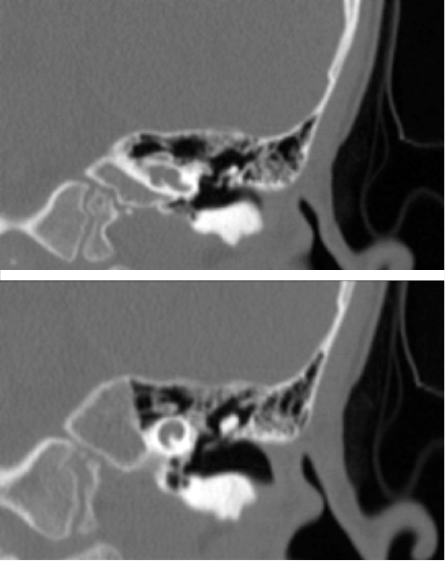
Figure 2: Coronal images of the left cochlea.
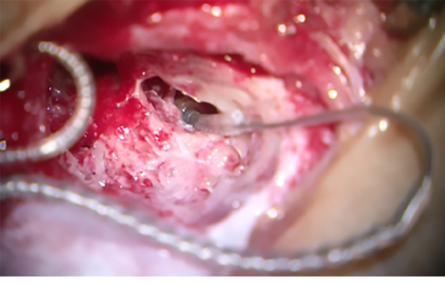
Figure 3: Posterior tympanotomy. Full insertion of the electrode in the basal turn of the right cochlea round window during the first operation.
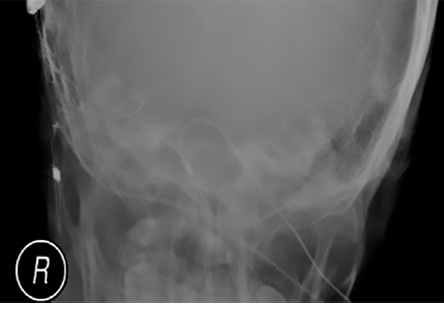
Figure 4: Postoperative X-ray demonstrating the misplaced cochlear implant electrode in the right internal auditory meatus.
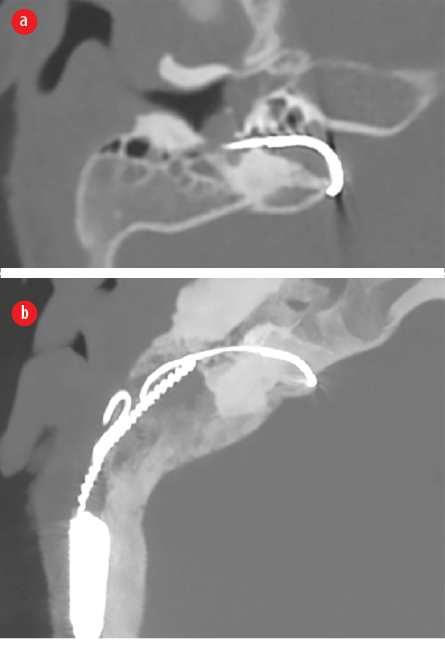
Figure 5: (a) Axial plane of internal auditory meatus (IAM) CT post-surgery showing the cochlear implant electrode passing through the IAM. (b) Maximum intensity projection reconstruction demonstrating the full course of the electrode from the subcutaneous device till the tip of the electrode reaching the intracranial fossa.
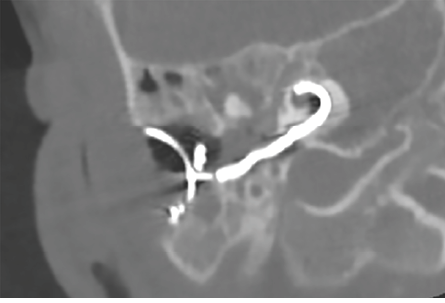
Figure 6: Coronal section of the right internal auditory meatus post repositioning of the electrode. The electrode is in place reaching the cochlear cavity.
Radiological imaging was performed to assess inner ear structures and to determine the selection criteria for surgical intervention.
Temporal bone HRCT was performed with a slice thickness of 0.8 mm. This revealed typical bilateral bony abnormalities affecting the inner ear structures but sparing the middle and external ear structures bilaterally. Both the IACs were bulbous in appearance with diameters of 7 mm on the left and 6.8 mm on the right [Figure 1a]. Normal diameter ranges from 2–8 mm with an average of 4 mm.2 The abnormalities included the symmetrical corkscrew appearance of the cochlea and absent bony separation between the basal turn of the cochlea and the IAC (lamina cribrosa) [Figure 1b]. In addition, poorly formed modiolus and interscalar septa were noted [Figure 2]. The facial nerve canal and vestibular aqueduct were prominent but were within normal limits.2,3 The semicircular canals were preserved. These CT features are considered characteristic of IP-III.
The child was given hearing aids, as per his parent’s preference, although cochlear implantation was advised. No improvement was noted following 18 months of regular follow-up and speech therapy, after which the family consented to proceed with cochlear implantation.
A perimodiolar electrode was selected following discussion with the otology committee accounting for the cochlear malformation and the short area of the basal turn to achieve full electrode insertion. Cochlear implantation was performed on the right side at the age of three years.
During surgery [Figure 3], the expected perilymph/cerebrospinal fluid (CSF) gushing occurred on exposing the round window, which gradually stopped after five minutes. The electrode of the implant was inserted and then sealed with periosteum to prevent fluid leakage.
Postoperative audiological tests showed some positive results in hearing; impedance field telemetry was detected, neural response telemetry (NRT) was satisfactory, and speech recognition threshold was seen in two channels.
However, abnormalities were identified on postoperative imaging. The X-ray revealed an abnormal straight course of the electrode [Figure 4], and a HRCT scan confirmed mispositioning of the electrode, the tip of which was reaching and hooking medially around the internal auditory meatus (IAM) [Figure 5].
The patient was taken back to surgery within 48 hours. The mispositioned cochlear implant was removed, and a new slim straight cochlear implant was re-implanted with great difficulty due to significant bleeding and perilymph/CSF fluid leak. Complete insertion could not be achieved; only 10 out of 24 channels could be inserted. The electrode position was confirmed by HRCT scanning immediately after surgery while the patient is still intubated [Figure 6] and the audiological NRT test demonstrated a satisfactory response.
The postoperative recovery period was uneventful, and follow-up audiological NRT testing demonstrated a normal response, representing a significant improvement compared to the initial test.
The child is under regular follow-up and, at five years of age, demonstrated normal attenuation of language milestones. No genetic testing was done for him.
Discussion
Genetic causes account for 50% of hearing loss; 70% of these are nonsyndromic, and less than 5% of the nonsyndromic hearing loss are X-linked. So far, five genes have been identified for X-linked nonsyndromic hearing loss.4 Cochlear IP-III is the rarest among the X-linked nonsyndromic causes of hearing loss. It is an X-linked recessive disorder caused by mutation of the POU3F4 gene located on chromosome X.1 It was first described genetically and clinically in 1971 by Nance et al.1,5 It is characterized by bilateral progressive mixed hearing loss, congenital fixation of the stapes, and a perilymph gusher (flow of perilymph/CSF into the middle ear when a stapedectomy or cochleostomy is attempted).5
Inner ear anomalies have been reported in 20% of patients with congenital hearing loss.4 According to the current literature, inner ear malformations are classified into eight distinct groups.6 IP-III anomaly represents around 4% of them.4
Recognition of these abnormalities can prevent unnecessary surgery, leading to perilymph gushing, and worsening of hearing loss. HRCT is the modality of choice for assessing congenital ear malformations with a recommended section thickness of 0.4–0.7 mm.2 It mainly illustrates the bony structures of the middle and internal ear. Magnetic resonance imaging is used preoperatively to assess the membranous labyrinth and the presence of normal nerves.5
X-linked anomaly radiological features were first described in the early 1970s by polytomography sections, which demonstrated widened IAC and absent bony plate separating IAC from the cochlea (lamina cribrosa).7,8 Phelps et al,9 was the first to report these anomalies on CT scan. He reported dilated bulbous IAC, incomplete separation of the cochlear basal turn and IAC, and dilated intratemporal parts of the facial canal. In addition to these findings, Talbot and Wilson subsequently described the absence of bony modiolus and abnormal vestibular aqueducts.10 Other features have been reported later in some patients, including structural abnormalities in the vestibule and the semicircular canals, small or atretic oval and round window,11 thickened stapes footplate, and hypertrophic Bill’s bar (a bony landmark that divides the superior compartment of the IAM into anterior and posterior compartments).12
The presence of bulbous IAC can be a normal variant in some individuals after excluding a tumor.9 However, when it coexists with the absent osseous separation between the lateral end of the IAC and basal turn of the cochlea, the direct communication between the two cavities forms what is called a corkscrew cochlea, a classical sign for cochlear IP-III.13 Moreover, this connection causes direct communication between CSF and perilymph, which increases the pressure in the cochlea and leads to gushing when the cochlea is opened during surgery.11
Despite the distorted architecture of the inner ear in these patients, the cochlea’s outer dimensions are normal, making cochlear implantation possible. In fact, it is the only means by which hearing can be improved in these patients. However, surgeons should be careful as these patients are at greater risk of developing complications. In addition to the commonly known complications of cochlear implantation, such as device failure and facial nerve stimulation, these patients can develop complications attributed to the absent bony septum separating the cochlear cavity from the IAC. The two main complications are CSF gushing into the middle ear during cochleostomy and misplacement of the cochlear implant electrode into the IAC. Position monitoring with intraoperative imaging is advised to prevent misplacement.14 Also, careful selection of the electrode type can minimize misplacement in these patients. Meningitis is also another possible complication. Covering the patient with vaccination and firm sealing of the cochleostomy to stop CSF leaks can prevent this risk.15
Conclusion
IP-III is one of the rarest inner ear anomalies. However, radiological features are typical for diagnosis. This should direct surgeons to avoid possible complications during stapes manipulation and cochlear implantation surgeries.
Disclosure
The authors declared no conflicts of interest. The patient gave consent to publish this case report.
references
- 1. Nance WE. The genetics of deafness. Ment Retard Dev Disabil Res Rev 2003;9(2):109-119.
- 2. Joshi VM, Navlekar SK, Kishore GR, Reddy KJ, Kumar EC. CT and MR imaging of the inner ear and brain in children with congenital sensorineural hearing loss. Radiographics 2012 May-Jun;32(3):683-698.
- 3. Gupta S, Mends F, Hagiwara M, Fatterpekar G, Roehm PC. Imaging the facial nerve: a contemporary review. Radiol Res Pract 2013;2013:248039.
- 4. Kanno A, Mutai H, Namba K, Morita N, Nakano A, Ogahara N, et al. Frequency and specific characteristics of the incomplete partition type III anomaly in children. Laryngoscope 2017 Jul;127(7):1663-1669.
- 5. Nance WE, Setleff R, McLeod A, Sweeney A, Cooper C, McConnell F. X-linked mixed deafness with congenital fixation of the stapedial footplate and perilymphatic gusher. Birth Defects Orig Artic Ser 1971 Mar;07(4):64-69.
- 6. Sennaroğlu L, Bajin MD. Classification and current management of inner ear malformations. Balkan Med J 2017 Sep;34(5):397-411.
- 7. Glasscock ME III. The stapes gusher. Arch Otolaryngol 1973 Aug;98(2):82-91.
- 8. Jensen J, Terkildsen K, Thomsen KA. Inner ear malformations with oto-liquorrhea. Tomographic findings in three cases with a mixed hearing impairment. Arch Otorhinolaryngol 1977 Jan;214(3):271-282.
- 9. Phelps PD, Reardon W, Pembrey M, Bellman S, Luxom L. X-linked deafness, stapes gushers and a distinctive defect of the inner ear. Neuroradiology 1991;33(4):326-330.
- 10. Talbot JM, Wilson DF. Computed tomographic diagnosis of X-linked congenital mixed deafness, fixation of the stapedial footplate, and perilymphatic gusher. Am J Otol 1994 Mar;15(2):177-182.
- 11. Incesulu A, Adapinar B, Kecik C. Cochlear implantation in cases with incomplete partition type III (X-linked anomaly). Eur Arch Otorhinolaryngol 2008 Nov;265(11):1425-1430.
- 12. Gong WX, Gong RZ, Zhao B. HRCT and MRI findings in X-linked non-syndromic deafness patients with a POU3F4 mutation. Int J Pediatr Otorhinolaryngol 2014 Oct;78(10):1756-1762.
- 13. Koontz NA, Seltman TA, Kralik SF, Mosier KM, Harnsberger HR. Classic signs in head and neck imaging. Clin Radiol 2016 Dec;71(12):1211-1222.
- 14. Sennaroğlu L, Bajin MD. Incomplete partition type III: A rare and difficult cochlear implant surgical indication. Auris Nasus Larynx 2018 Feb;45(1):26-32.
- 15. Özcan M, Oğuzhan R, Baklacı D, Dündar G, Yılmaz F, Toprak U, et al. Cochlear implantation: case report on incomplete partition type III. Kulak Burun Bogaz Ihtis Derg 2017;27(3):146-150 .