A 36-year-old male presented with multiple red spots over the tongue that appeared at the age of 10 years. In the past two decades, he had suffered from recurrent spontaneous episodes of profuse bleeding from the nose and tongue lesions, requiring hospitalization with multiple blood transfusions. He also complained of exertional breathlessness, easy fatigability, palpitations, and frequent blackouts for the same duration. There was no associated bleeding gums, abdominal pain, melena, seizures, visual disturbances, or hemoptysis. His recently deceased elder brother (in a road traffic accident) had similar mucocutaneous lesions and a history of occasional nose bleeding since adolescence. Their father, too, had non-traumatic epistaxis episodes and died at a young age due to severe hematemesis.
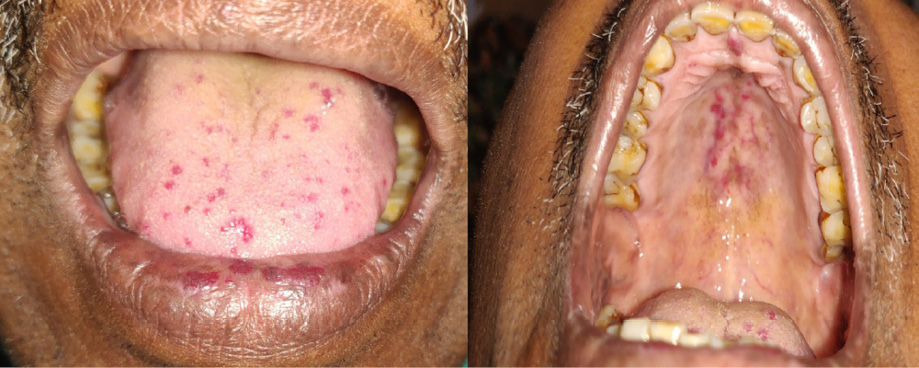
Figure 1: Multiple erythematous papulonodules over the tongue, lower lip, and palate in a 36-year-old male.
Clinical examination revealed pallor, multiple erythematous compressible non-pulsatile papulonodules over the tongue, and multiple petechiae over the palate [Figure 1]. Laboratory investigations were notable for severe anemia. Upper gastrointestinal endoscopy revealed multiple petechiae in the intestinal mucosa. Imaging studies of the brain, thorax and abdomen, and colonoscopy were non-contributory.
Question
- What is the most likely diagnosis?
a. Ataxia telangiectasia.
b. Unilateral nevoid telangiectasia.
c. Fabry disease.
d. Osler-Weber-Rendu syndrome.
e. Microcystic lymphangioma.
Answer
D. Osler-Weber-Rendu syndrome.
The above findings fulfill the diagnostic criteria of Osler-Weber-Rendu syndrome (OWRS). The patient received a blood transfusion, intravenous iron supplementation, and endoscopic cauterization for gastrointestinal arteriovenous malformation (AVM). He has been followed up at regular intervals for the past year without any sequelae.
Discussion
OWRS, also known as hereditary hemorrhagic telangiectasia, is an autosomal dominant disorder clinically characterized by mucocutaneous and gastrointestinal telangiectasia, spontaneous recurrent epistaxis, and multiorgan AVM. Fulfillment of three out of four criteria of the Curaçao criteria (visceral AVMs, mucocutaneous telangiectasias, spontaneous and recurrent epistaxis, and positive family history) warrants a definitive diagnosis of this condition.1 Recurrent epistaxis episodes may be the initial presentation of the disease during childhood. The characteristic mat like and papular telangiectasias (small tufts of dilated capillary scattered over the mucous membranes) appear during adolescence. Telangiectasias tend to increase in size and number with progressive age and are most commonly seen over the face, tongue, lips, nasal mucosa, hands, and fingertips. The gastrointestinal tract is frequently affected resulting in obvious hematemesis, melena culminating into iron deficiency anemia. Chronic persistent anemia requiring iron and blood transfusion is characteristic of severe disease, as encountered in our case. Pulmonary and cerebral AVM may appear later in life. Other sites of visceral bleeding may be the kidney, spleen, liver, meninges, and brain.2
Ataxia-telangiectasia is an autosomal recessive disorder characterized by cerebellar ataxia, growth retardation, chest infections, immunodeficiency, and oculocutaneous telangiectasia (oral cavity, malar area, ears, and eyelids). Telangiectasias in unilateral nevoid telangiectasia are usually confined to the trigeminal and upper cervical dermatomes, often associated with situations of relative estrogen excess. Angiokeratomas (punctate dark-red to blue-black papules with a slight keratotic surface that do not blanch on pressure) that characterize Fabry disease will be accompanied by additional features like pain and paresthesias of the extremities, hypohidrosis, and renal and coronary insufficiency. Clusters of translucent vesicles resembling frogspawn typify lymphangioma circumscriptum, a benign lymphatic malformation. Hemorrhages and leakage of superficial lymph within the vesicles can mimic the clinical picture, but accompanying systemic findings will not be elicited.3
Treatment is directed at controlling the specific complications and identifying and treating the AVMs before symptom-onset. Estrogen therapy, thalidomide, bevacizumab, cryotherapy, diathermy, cautery, or laser may be used to treat individual lesions.4 Surgical management or embolization may be required for uncontrolled bleeding from mucosal lesions or visceral lesions.
Any patient presenting with multiple telangiectasias and frequent nasal bleeds requires prompt and thorough systemic evaluation to look for visceral AVMs and subsequent management to reduce the disease’s morbidity and mortality.
Disclosure
The authors declared no conflicts of interest.
Acknowledgements
The authors would like to thank the patient for his consent.
references
- 1. Guttmacher AE, Marchuk DA, White RI Jr. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995 Oct;333(14):918-924.
- 2. Thappa DM, Laxmisha C. Recurrent bleeding from the tip of index finger and mucocutaneous telangiectases. Osler-Weber-Rendu syndrome. J Postgrad Med 2002 Oct-Dec;48(4):274-287.
- 3. James WD, Berger TG, Elston DM, Neuhaus IM. Andrews’ diseases of the skin clinical dermatology. 12th ed. Philadelphia:Elsevier;2016.
- 4. Harries PG, Brockbank MJ, Shakespeare PG, Carruth JA. Treatment of hereditary haemorrhagic telangiectasia by the pulsed dye laser. J Laryngol Otol 1997 Nov;111(11):1038-1041.