Thyroid hormones (THs), thyroxine (T4) and triiodothyronine (T3), regulate skeletal growth, maturation of the central nervous system, energy homeostasis, positive chronotropic and inotropic cardiac effects, and gastrointestinal functions. The hypothalamic pituitary thyroid axis accomplishes this process. These physiological effects are mediated through TH nuclear receptors (TR),1 which have been classified into α and β receptors.
TRα (TRα1, TRα2) and TRβ (TRβ1, TRβ2) have different tissue distributions.TRα1 is the predominant subtype in bone, the gastrointestinal tract, central nervous system, and cardiac and skeletal muscles, while TRα2 location and role are not yet clear. TRβ1 is the primary subtype in the liver and kidney, while TRβ2 dominates in the hypothalamus, pituitary, cochlea, and retina.2–4
Various genetic conditions of partial insensitivity to THs have been described. The resulting defects can cause either an alteration of the β or α nuclear receptors, a disruption of an intracellular transporter (monocarboxylate transporter 8), or a defect in their activation in the peripheral tissues due to failed activation of deiodinases by selenoproteins (selenocysteine insertion sequence binding protein 2).5
The reported incidence of resistance to thyroid hormon beta (RTHβ), which is a dominantly inherited disorder due to TRβ gene mutations is 1 in 40 000.6 Most patients with RTHβ are clinically euthyroid, but they can present with various symptoms and signs as summarized in Table 1. Biochemically, they manifest with high serum free T4 (fT4), fT3, and thyroid-stimulating hormone (TSH).7
However, clinical features of resistance to thyroid hormone alpha ((RTHα) (OMIM 614450) which was first reported in 2012, including dysmorphic facies, skeletal dysplasia (macrocephaly, epiphyseal dysgenesis), growth retardation, constipation, dyspraxia, and intellectual deficit.
Table 1: Comparison between the clinical features of resistance to thyroid hormone β (RTHβ) versus resistance to thyroid hormone α (RTHα).
|
Clinical features |
|
|
|
Thyroid gland |
Goiter |
No goiter |
|
Dysmorphism |
Nil |
Dysmorphic facies |
|
Skeletal changes |
Nil |
Macrocephaly, epiphyseal dysgenesis, Wormian bones |
|
Hematological |
Nil |
Anemia |
|
Heart |
Tachycardia |
Bradycardia |
|
Nervous system and behavior |
Emotional disturbances, hyperkinetic behavior, attention deficit hyperactivity disorder, learning disability, mental retardation
(intelligence quotient < 70) |
Autistic spectrum disorder, dyspraxia, learning disability, intellectual deficit |
|
Growth and development |
Short stature (< 5%), delayed bone age > 2 standard deviation, low body mass index (in children), hearing loss (sensorineural) |
Short stature, delayed bone age, high body mass index, delayed mile stones |
|
Gastrointestinal |
|
Chronic constipation |
|
Recurrent ear and throat
Infections |
Yes, 55% |
No |
|
Biochemical profile |
|
|
|
free T4 |
High |
Low/low-normal |
|
free T3 |
High |
High/high-normal |
|
Thyroid-stimulating hormone |
High |
Normal |
|
Reverse T3 |
- |
Low |
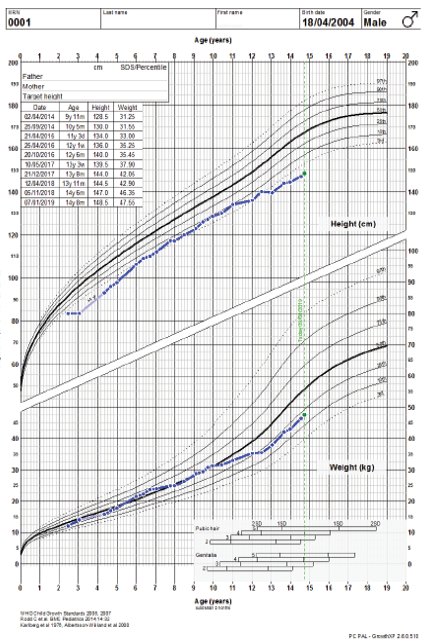
Figure 1: Growth charts of the patient.
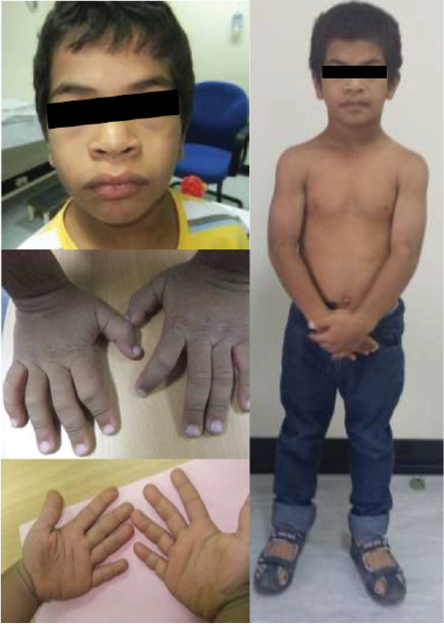
Figure 2: The patient aged 10. Note the characteristic facial features such as macrocephaly, coarse and flat face, periorbital fullness, hypertelorism, flat broad nasal bridge, upturned nose, anteverted nares, thick prominent lips, generalized muscular hypertrophy, and puffy, short, broad hands with rough and doughy texture skin.
The biochemical features are typically borderline normal thyroid function tests with low/low-normal fT4 and high/high-normal fT3 concentrations, a subnormal fT4/fT3 ratio, reduced reverse T3, raised muscle creatinine kinase, and mild anemia.8
To date, 31 patients have been reported in the literature carrying 20 missense and frameshift mutations.9,10 None of the described patients are from Arab countries. The authors searched the EMBASE, Medline, and PubMed databases using ‘thyroid hormone resistance alpha’ as search words, and none were reported from the Middle East and North Africa (MENA) region.
Case report
A 13-year-old male was referred at the age of three months to our center for evaluation of anemia. He was found to have hypochromic microcytic anemia diagnosed as iron deficiency anemia. However, his anemia persisted after correction of the iron deficiency by a therapeutic dose of ferrous sulfate, evidenced by persistent normochromic normocytic anemia and normal bone marrow iron stores. Reduced activity, excessive sleepiness, and chronic constipation were also reported since early infancy. The child was on regular follow-up with a hematologist; subtle dysmorphism was reported. Later, during his follow-up, his growth was suboptimal, and there was a delay in achieving the age-appropriate developmental milestones.
His developmental delay manifested as an impairment of his gross motor, coordination, language, and cognitive abilities. He started to walk independently at the age of eighteen months but with imbalance. His speech was slow and monotonous, with no major receptive or expressive deficit. His school performance was suboptimal in almost all subjects and required extra teaching sessions.
The child’s growth was slow from two and a half years of age. At the age of three years, his height was 83 cm below the third percentile (-3.5 standard deviation (SD)) for his age. He was obese, weight for age was 14 kg at 41st percentile (-0.2 SD), and he had a body mass index of 20.1 kg/m2 above the 99th percentile (+3.2 SD). He had disproportionate short stature, with an upper/lower segment ratio > 1.7, predominantly affecting the lower segment. Furthermore, he had macrocephaly; his head circumference was > 97th percentile (+2.0 SD) [Figure 1]. He had thick dry myxedematous skin with generalized hypertrichosis and muscle hypertrophy. Dysmorphic features including coarse facial features, low anterior and posterior hairline, periorbital fullness, short palpebral fissure, telecanthus and hypertelorism, epicanthic folds, broad and flat nasal bridge, midface hypoplasia, splayed out nasal base with flat nasal tip, anteverted nares, thick prominent lips, low set ears with thick and prominent ear lobes, brachydactyly, and broad hands [Figure 2]. He has a mild increase in joint laxity. His teeth were small and widely spaced with multiple dental caries. He had generalized muscular weakness with a wide-based and waddling gait. Gower’s sign was negative. Muscle tone and power were normal with brisk reflexes.
The child has been investigated and seen by different specialties, including a hematologist, general pediatrician, endocrinologist, neurologist, geneticist, cardiologist, developmental pediatrics specialist, and speech and occupational therapists.
The child was thoroughly investigated, and his initial workup showed a microcytic, hypochromic anemia. He was evaluated for inborn errors of metabolism. His urine screening was negative for glycosaminoglycans and oligosaccharides. Skeletal survey showed that both femoral epiphysis were small with shallow acetabulum and minimal anterior beaking of lumbar vertebrae. Genetic workup showed a normal karyotype, with normal array comparative genomic hybridization and negative fluorescence in situ hybridization for 7q11.23 microdeletion which ruled out Williams Beuren syndrome (OMIM 194050). Sequencing for polymerase I and transcript release factor mutation for congenital generalized lipodystrophy type 4 (CGL4; OMIM 61337) was also negative.
He had repeated normal levels of fT4 and TSH, persistent mild elevation of creatine kinase (CK) around 788 U/L (39–308), low insulin-like growth factor (IGF1) 68 µg/L (115–498), with failure to respond to two growth hormone (GH) stimulation tests as the peak of provoked growth hormone was only 15 mIU/L (our lab’s cut off is 20 mIU/L). We opted for the “wait and see” approach initially, and then he was started on GH therapy after four years of age. He initially showed a good response but later went suboptimal, which was thought to be due to poor adherence to GH therapy and/or the underlying pathology related to the genetic mutation. His height before GH therapy was -2.9 SDs, which gradually improved to -1.4 SD at 11 years old but later deteriorated to -2.4 SD [Figure 1].
Skeletal radiographic study showed delayed bone age by two years at chronologic age of six years. His neurological evaluation showed normal brain magnetic resonance imaging (MRI). MRI of thigh muscle showed only muscular hypertrophy. His muscle biopsy showed normal morphology with a negative caveolin stain. Nerve conduction study showed bilateral delay in both motor and sensory divisions of his median nerve. An echocardiogram showed mild left ventricular trabeculations. His brainstem auditory evoked potentials were normal. His IQ assessment was found to be below-average intelligence (IQ 87). Although initial THs levels were normal, subsequent measurements revealed persistently elevated levels of fT3 and normal levels of fT4 and TSH [Table 2].
Table 2: Biochemical profile of the patient before and during treatment.
|
Dose of Levothyroxine, µg |
|
|
|
|
25 |
50 |
75 |
100 |
|
Free T4 (9.7–14.2 pmol/L) |
8.8 |
8.7 |
11.1 |
12.2 |
13.3 |
14.3 |
18.3 |
10.9 |
|
Free T3 (3.8–6 pmol/L) |
7.6 |
|
|
7.8 |
8.0 |
8.1 |
9.6 |
7.9 |
|
TSH (0.8–3.9 mIU/L) |
2.70 |
2.30 |
1.66 |
1.68 |
1.42 |
0.47 |
0.01 |
0.02 |
|
Free T4:Free T3 (2.0–4.5 pmol/L) |
1.2 |
|
|
1.6 |
|
|
|
|
|
CK (39–308 U/L) |
|
713 |
327 |
264 |
|
|
|
|
|
CK, Mb |
|
|
19.4 |
|
|
|
|
|
|
Hb (11.5–15.5 g/dL) |
9.6 |
9.5 |
10.3 |
|
|
|
|
10.9 |
|
MCV (78.0–96.0 fL) |
71.8 |
72.7 |
77.3 |
|
|
|
|
78.0 |
|
MCH (26.0–33.0 pg) |
23.7 |
24.5 |
25.0 |
|
|
|
|
23.7 |
|
MCHC (31.0–35.0 g/L) |
33.1 |
33.7 |
32.4 |
|
|
|
|
30.4 |
TSH: thyroid-stimulating hormone; CK: creatine kinase; Hb: hemoglobin; MCV: mean corpuscular volume; MCH: mean corpuscular hemoglobin; MCHC: mean corpuscular hemoglobin concentration; IGFI: low insulin-like growth factor.
In 2016, TRIO whole-exome sequencing detected that the patient was heterozygous for a novel, de novo mutation in the TH receptor α (THRA) (NM-001190910) gene, (c.871G>A, p.Gly291Ser), which was absent in his parents.
Our institutional ethics committee approved the genetic study. The patient’s parents provided written informed consent. We performed whole-exome sequencing for the patient and his parents. Bioinformatics analysis of the sequence data identified a novel de novo mutation that is linked with the patient’s phenotype.
This is the first case report from Oman and the MENA region. At the age of 12, he was started on thyroxine treatment with gradual dose increment from 25 µg to 100 mcg with monitoring of his clinical and biochemical response. Close monitoring of his biochemical indices showed suppression of TSH. His fT3 level remained in the supra-physiological range. He showed some clinical response in terms of improved general wellbeing and some improvement in constipation. He went into normal puberty. He still suffers from significant learning difficulties. However, he continued attending a mainstream school with inclusion classes.
Discussion
RTHβ is extensively studied, however, it is likely that RTHα is more common than expected and may even be more common than RTHβ. The difference in the presentation is most likely due to the differences in tissue expression of TRα and TRβ.11 Recently, several patients with RTHα due to THRA mutations were identified. These patients’ phenotype consists of varying degrees of growth impairment, delayed bone, mental and motor development, constipation, macrocephaly, and near-normal thyroid function tests.7,12–18 The phenotype of mice harboring different TRα mutations is not uniform, suggesting that the human disorder may also be variable.19
Our patient had many clinical features typical of congenital hypothyroidism, but that paradoxically was associated with normal levels of fT4 and TSH with elevated fT3 levels and low fT4/fT3 ratio. He had normochromic normocytic anemia and developmental delay; mainly impairing his gross motor coordination, language, and cognitive abilities. He also had disproportionate short stature with femoral epiphyseal dysgenesis, and delayed bone age, coarse facial features and generalized muscular hypertrophy with high CK (Kocher–Debré–Semelaigne Syndrome).
Molecular genetic study identified a novel heterozygous mutation in the THRA in a highly conserved amino acid that is likely to be the cause, which describes the phenotype and the endocrinopathy the patient has.
The patient was treated using GH therapy, for which he showed a good response in improving his linear growth with delta of +1.5 SD. Although treatment with thyroxine started late, marked improvement in his general wellbeing and some improvement in constipation noted, but that was not reflected on the linear growth or reducing the skin thickness, or minimizing the coarse facial features. Biochemically, starting thyroxine increased fT4, suppressed TSH, and maintained fT3 at supra-physiological levels. Furthermore, his anemia persisted. Reviewing the literature, the benefit of thyroxine replacement in patients with RTHα is not as effective but showed some benefits, especially if started early in life. In three patients described by Moran et al,15 starting thyroxine between the age of two to three years improved their resting energy expenditure with a subjective feeling of being more energetic. It also improved constipation, and growth and development. Furthermore, a case from the Netherlands treated at 18 months of age showed a marked improvement in growth and development over a period of 12 months.10 Other reported patients had no improvement, which may be related to the severity of the structural alteration of the causative mutant TRα1 so that the more severe the alteration, the less effective the thyroxine therapy.20 It may also reflect the delay in initiating thyroxine treatment. Researchers have started thinking about alternative therapy for this cohort of patients. Histone deacetylases inhibitor like suberoylanilide hydroxamic acid was tried on mice model, but it showed no benefit on skeletal development.21 Other types of therapies might emerge soon as the discovery of this mutation had attracted the attention of researchers worldwide.
Our case highlights the need to maintain a high index of suspicion when the phenotype suggests hypothyroidism but with normal fT4 and TSH. The measurement of the levels of fT3 can help to reach the diagnosis of RTHα.
Conclusion
Although it was thought that RTHα is rare, improvements in molecular genetic testing have led to the discovery of more cases. TRα resistance secondary to a mutation in the THRA gene has distinct clinical features along with physiological features of hypothyroidism in specific tissues. Almost all patients will have subtle or no abnormalities in fT4. These cases are not usually detected by newborn screening, as they do not manifest with high TSH. It is important to consider resistance to THα in children with refractory anemia, severe constipation, short stature, and subtle dysmorphism. Although many cases present with symptoms because of the subtle changes in the thyroid function tests, many patients remain undiagnosed for a long time, even in tertiary hospitals. In such cases, in addition to fT4 and TSH, other investigations, including fT3, CK should be considered. Clinical dysmorphology assessment and genetic testing is indicated. Thyroxine therapy can reverse some clinical and/or biochemical abnormalities. However, careful monitoring for excessive TH adverse effects in hormone-sensitive TRβ tissues is recommended. Delayed treatment can result in lower neurocognitive outcomes.
Disclosure
The authors declared no conflicts of interest.
references
- 1. Mullur R, Liu YY, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev 2014 Apr;94(2):355-382.
- 2. Lazar MA. Thyroid hormone receptors: multiple forms, multiple possibilities. Endocr Rev 1993 Apr;14(2):184-193.
- 3. Yen PM. Physiological and molecular basis of thyroid hormone action. Physiol Rev 2001 Jul;81(3):1097-1142.
- 4. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev 2010 Apr;31(2):139-170.
- 5. Refetoff S, Bassett JH, Beck-Peccoz P, Bernal J, Brent G, Chatterjee K, et al. Classification and proposed nomenclature for inherited defects of thyroid hormone action, cell transport, and metabolism. Eur Thyroid J 2014 Mar;3(1):7-9.
- 6. Lafranchi SH, Snyder DB, Sesser DE, Skeels MR, Singh N, Brent GA, et al. Follow-up of newborns with elevated screening T4 concentrations. J Pediatr 2003 Sep;143(3):296-301.
- 7. Refetoff S, Weiss RE, Usala SJ. The syndromes of resistance to thyroid hormone. Endocr Rev 1993 Jun;14(3):348-399.
- 8. van Mullem A, van Heerebeek R, Chrysis D, Visser E, Medici M, Andrikoula M, et al. Clinical phenotype and mutant TRα1. N Engl J Med 2012 Apr;366(15):1451-1453.
- 9. Sun H, Wu H, Xie R, Wang F, Chen T, Chen X, et al. New case of thyroid hormone resistance α caused by a mutation of THRA/TRα1. J Endocr Soc 2019 Feb;3(3):665-669.
- 10. van Gucht AL, Meima ME, Zwaveling-Soonawala N, Visser WE, Fliers E, Wennink JM, et al. Resistance to thyroid hormone alpha in an 18-month-old girl: clinical, therapeutic, and molecular characteristics. Thyroid 2016 Mar;26(3):338-346.
- 11. Visser WE, Friesema EC, Visser TJ. Minireview: thyroid hormone transporters: the knowns and the unknowns. Mol Endocrinol 2011 Jan;25(1):1-14.
- 12. Bochukova E, Schoenmakers N, Agostini M, Schoenmakers E, Rajanayagam O, Keogh JM, et al. A mutation in the thyroid hormone receptor alpha gene. N Engl J Med 2012 Jan;366(3):243-249.
- 13. Moran C, Schoenmakers N, Agostini M, Schoenmakers E, Offiah A, Kydd A, et al. An adult female with resistance to thyroid hormone mediated by defective thyroid hormone receptor α. J Clin Endocrinol Metab 2013 Nov;98(11):4254-4261.
- 14. van Mullem AA, Chrysis D, Eythimiadou A, Chroni E, Tsatsoulis A, de Rijke YB, et al. Clinical phenotype of a new type of thyroid hormone resistance caused by a mutation of the TRα1 receptor: consequences of LT4 treatment. J Clin Endocrinol Metab 2013 Jul;98(7):3029-3038.
- 15. Moran C, Agostini M, Visser WE, Schoenmakers E, Schoenmakers N, Offiah AC, et al. Resistance to thyroid hormone caused by a mutation in thyroid hormone receptor (TR)α1 and TRα2: clinical, biochemical, and genetic analyses of three related patients. Lancet Diabetes Endocrinol 2014 Aug;2(8):619-626.
- 16. Tylki-Szymańska A, Acuna-Hidalgo R, Krajewska-Walasek M, Lecka-Ambroziak A, Steehouwer M, Gilissen C, et al. Thyroid hormone resistance syndrome due to mutations in the thyroid hormone receptor α gene (THRA). J Med Genet 2015 May;52(5):312-316.
- 17. Espiard S, Savagner F, Flamant F, Vlaeminck-Guillem V, Guyot R, Munier M, et al. A novel mutation in THRA gene associated with an atypical phenotype of resistance to thyroid hormone. J Clin Endocrinol Metab 2015 Aug;100(8):2841-2848.
- 18. Demir K, van Gucht AL, Büyükinan M, Çatlı G, Ayhan Y, Baş VN, et al. Diverse genotypes and phenotypes of three novel thyroid hormone receptor-α mutations. J Clin Endocrinol Metab 2016 Aug;101(8):2945-2954.
- 19. Vennström B, Mittag J, Wallis K. Severe psychomotor and metabolic damages caused by a mutant thyroid hormone receptor alpha 1 in mice: can patients with a similar mutation be found and treated? Acta Paediatr 2008 Dec;97(12):1605-1610.
- 20. Tang Y, Yu M, Lian X. Resistance to thyroid hormone α, revelation of basic study to clinical consequences. J Pediatr Endocrinol Metab 2016 May;29(5):511-522.
- 21. Freudenthal B, Shetty S, Butterfield NC, Logan JG, Han CR, Zhu X, et al. Genetic and pharmacological targeting of transcriptional repression in resistance to thyroid hormone alpha. Thyroid 2019 May;29(5):726-734.