Pheochromocytomas are tumors derived from chromaffin cells within the medulla of the adrenal glands, whereas paragangliomas are tumors derived from extra-adrenal chromaffin cells of the sympathetic prevertebral and paravertebral ganglia. Paragangliomas can occur anywhere in the sympathetic system. They predominantly occur in the chest, abdomen, and pelvic regions but can also arise from parasympathetic ganglia located in the neck and at the base of the skull.1 Pheochromocytoma and sympathetic paraganglioma usually secrete considerable amounts of catecholamines such as epinephrine, norepinephrine and/or dopamine, causing symptoms of catecholamine excess such as blood pressure, headache, intermittent sweating episodes, strong palpitations, and anxiety. Conversely, parasympathetic paragangliomas are mostly non-functioning and are generally detected after tumor-mass growth.1 The risk for developing metastatic disease is greater for extra-adrenal sympathetic paragangliomas than for pheochromocytomas.
The growing deployment of genetic testing has shown that approximately 30% of pheochromocytoma and paraganglioma (PPGL) patients carry familial pathogenic germline mutations in known PPGL-susceptibility genes.2 This generally leads to genetic screening of their family members as well. As a result, there is an increase in the detection of asymptomatic PPGLs or non-secreting tumors reported in familial cases discovered after index patient work-up. Genetic screening has thus enabled the early detection of PPGLs, for which early surgical intervention can significantly impact prognosis and treatment strategies to reduce morbidity and mortality.
The main challenges for the management of hereditary PPGL syndromes include decisions on regions for surveillance, the timing of screening, and the need/timing of surgery. Although PPGLs are similar tumors, they warrant distinction from each other concerning their differences in locations, manifestations, secretory functions, genetic syndromes, and propensity to metastasize.
This case report presents an Omani family with a history of paraganglioma and highly variable clinical presentations among the affected members. This report highlights the complexity of managing such families and encourages further discussion regarding the management of asymptomatic PPGL-associated mutation carriers.
Case report
Case one
Our index patient was a 14-year-old Omani boy [Figure 1, patient II.15, aged 20 years old at present] from a remote village in Oman. He was referred to our clinic in 2012 with hypertension and extra-adrenal lesions detected by computed tomography (CT) scan in the left posterior mediastinum and the right pelvic region near the urinary bladder. The index patient was thinly-built with a body mass index of 18 kg/m2. Blood pressure was 170/100 mmHg despite being on the maximum dose of the calcium channel blocker, nifedipine. His blood pressure control improved after adding a selective alpha-blocker in combination with nifedipine (the biochemical profile is shown in Table 1). A whole-body magnetic resonance imaging ( MRI) scan of patient II.15 revealed a 7.2 cm × 5.2 cm × 4.4 cm mass at the left paraspinal area extending from T8/T9 to T10/T11 level [Figure 2a] and a right pelvic mass approximately 3.5 cm × 3.2 cm closely related to the right external and internal iliac vessels consistent with left paraspinal and right pelvic paraganglioma [Figure 2b], respectively. The functional imaging iodine-123 metaiodobenzylguanidine (I-131MIBG) scan indicated an MIBG-avid tumor in the lower chest with an equivocal area near the bladder [Figure 2c]. The patient underwent left thoracotomy and laparotomy with resection of both masses. Histopathology analyses of both lesions were consistent with paraganglioma and immunohistochemistry showed positive staining for Chromogranin A (CgA) and synaptophysin (S-100) in sustentacular cells [Figure 2d and e, respectively]. The early-age onset of multifocal disease presentation in patient II.15 warranted his referral for genetic testing after obtaining informed consent from his parents. This revealed a heterozygous pathogenic succinate dehydrogenase B (SDHB) mutation:c.574T>C; p.Cys192Arg (coding reference: NM_00300.2). As patients with mutations in the SDHB gene have a high risk of other malignancy like papillary thyroid cancer, a neck ultrasound was performed in the patient, which was normal. His blood pressure normalized after surgery. His initial and annual follow-up with CgA and plasma metanephrine levels were normal.
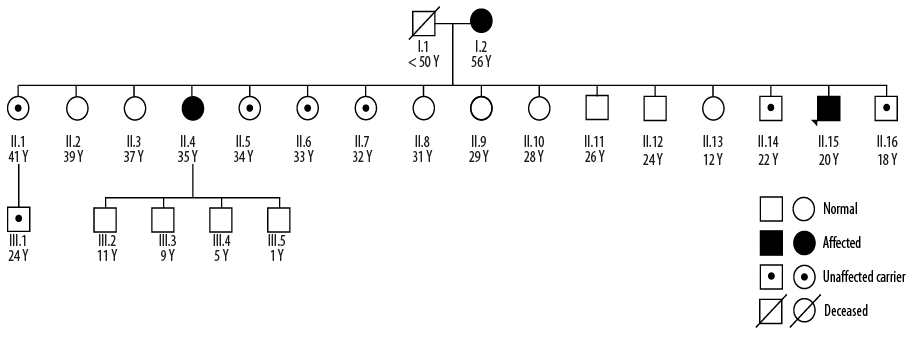
Figure 1: Family pedigree of an Omani family with a history of paraganglioma. Patient II.15 was a 14-year-old male (currently 20 years old) who was the index case from this family. Patient II.4 is his sister who presented at the age of 35, and Patient I.2 is their 56-year-old mother. The father of the index patient died of unknown reasons. Eight out of the 16 siblings (50.0%) in generation II were carriers of the succinate dehydrogenase B:c.574T>C mutation, reflecting the autosomal dominant inheritance risk for paraganglioma and other associated tumors.
Subsequently, the asymptomatic 15 siblings and mother of the index patient were screened for the SDHB:c.574T>C mutation. The father of the index patient had died at a young age (< 50 years) from an unknown cause. The SDHB:c.574T>C mutation was identified in the mother, seven siblings, and one nephew of the index patient. All the mutation-positive individuals of this family were recruited for annual biochemical screening with CgA and plasma metanephrine and normetanephrine. This led to the detection of our next case.
Table 1: Biochemical profile of the index patient, Patient II.15.
|
RFT, TFT |
Normal |
|
|
Urine normetanephrine |
28 150 nmol/L |
< 3300 nmol/L |
|
Urine metanephrine |
530 nmol/L |
< 1800 nmol/L |
|
Plasma metanephrine |
0.28 nmol /L |
0–0.5 nmol/L |
|
Plasma normetanephrine |
1300 nmol/L |
0–1 mol/L |
RFT: renal function test; TFT: thyroid function test; CgA: chromogranin A.
Case two
Except for the 34-year-old sister [Figure 1, Patient II.4, 35 years old at present] of the index patient, all mutation carriers screened were biochemically silent. At the time of genetic screening, Patient II.4 was a mother of three children with uneventful previous pregnancies. Her clinical parameters were unremarkable. Her biochemical screening results showed high serum normetanephrine 2.3 nmol/L (normal: < 0.1 nmol), a high CgA 209 µg/L (normal: 27–92 µg/L), and normal metanephrine level 0.2 nmol/L (normal: 0–0.5 nmol) which indicates a unsuspected paraganglioma. Patient II.4 was immediately referred for a whole-body CT scan. However, she turned out to be in her initial month of her fourth pregnancy at the time of the appointment. Eventually, a whole-body MRI scan without gadolinium contrast at 30 weeks of gestation was arranged after informed consent regarding high risk. The MRI revealed pre- and paravertebral soft tissue mass extending from the level of inferior mesenteric artery and down to the pelvic region spanning over 11.5 cm length in Patient II.4. The mass partially encased the lower part of the aorta, and no enlarged mesenteric lymph nodes were observed. Her antenatal care was transferred under a multidisciplinary team. At 32 weeks of gestation, her blood pressure started to rise, and she was managed with labetalol and nifedipine due to intolerance to prazosin.
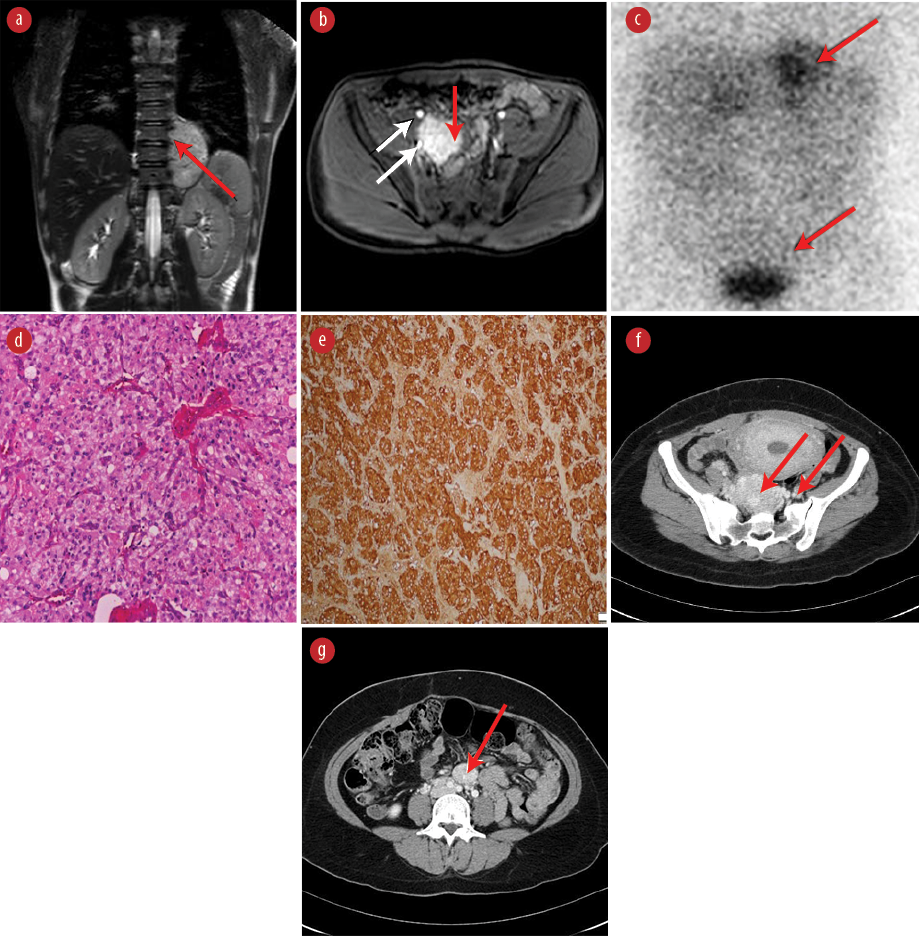
Figure 2: Scan and pathology results of paragangliomas in patient II.15 (a-e) and patient II.4 (f and g). (a) Weighted coronal image reveals a 7.2 cm × 5.2 cm × 4.4 cm mass of moderate hyperintensity in the left paraspinal area extending from T8/T9 to T10/T11 level (red arrow). (b) T1-weighted postcontrast image, showing a right pelvic mass, approximately 3.5 cm × 3.2 cm (red arrow) situated between the right external and internal iliac vessels (white arrows), showing intense homogeneous enhancement. (c) MIBG scan shows an increased uptake in the left mediastinal and right pelvic region (red arrows) corresponding to the anatomical tumor locations on MRI scan. (d) Hematoxylin and eosin stained tumor tissue shows well-circumscribed nodule composed of nets of monomorphic cells having abundant eosinophilic cytoplasm and coarse chromatin, consistent with paraganglioma. (e) Immunohistostaining showing positive staining for Chromogranin A and synaptophysin in sustentacular cells. (f) An axial CT scan with intravenous contrast showing a large mass in the left para-aortic region with intense and homogenous enhancement (red arrows) in favor of paraganglioma of Zuckerkandl. (g) Axial CT scan with contrast showing a large mass with intense and homogenous enhancement (red arrow) at S3 level consistent with pelvic paraganglioma.
At 37 weeks of gestation, elective cesarean section was carried out uneventfully. Three months after delivery, a whole-body CT scan with contrast revealed three separate lesions in Patient II.4. A retroperitoneal pre- and paravertebral soft tissue-density lesion was noted, extending from the level of L4 to the S1 vertebrae measuring 10.5 cm × 5.3 cm × 3.3 cm starting below the aortic bifurcation and body of Zuckerkandl [Figure 2f]. A similar lesion measuring 2.7 cm × 2.2 cm was observed anterior to the aortic bifurcation [Figure 2g]. Another small lesion measuring 1.2 cm × 1.1 cm was detected at the medial aspect of the left adrenal gland. Subsequently, she underwent laparoscopic left adrenalectomy and excision of the left para-aortic paraganglioma and laparotomy and excision of the large retroperitoneal paraganglioma. Histopathological analysis revealed left adrenal pheochromocytoma and multiple paraganglioma with negative mesenteric lymph nodes. Her follow-up at three weeks, and three and six months after surgery showed normal serum normetanephrine and CgA levels. An Octreoscan showed no evidence of local recurrence or distant metastases. Currently, she is off medication and normotensive. Her four children tested negative for the SDHB:c.574T>C mutation.
Based on the Endocrine Society guidelines,3 whole-body CT scans were ordered for all other mutation-positive family members. This led to our third case from this family.
Case three
A whole-body CT scan identified a 3 cm middle mediastinal mass and a few nodules (largest lesion measured 18 m × 13 mm × 11 mm) in the thyroid gland of the 56-year-old mother of the index patient [Figure 1, Patient I.2]. She had no significant medical or surgical history. Ultrasound and fine-needle aspiration cytology indicated that the thyroid lesions were benign. She was further assessed using Octreoscan, and the result was consistent with middle mediastinal paraganglioma with rest of the body being normal. The patient is currently awaiting surgery.
Discussion
PPGLs constitute of a group of rare disorders in Oman, with fewer than 10 patients referred annually to our National Endocrine Center. However, the increase in genetic screening and follow-up surveillance of PPGL patients and their family members have raised the need for guidelines, particularly for asymptomatic carriers.
Here, we present case reports of three affected members of a single Omani family with a history of paraganglioma and highly variable clinical presentations among affected members. Eight out of the 16 siblings (50.0%) in the second generation of the reported family pedigree [Figure 1] were carriers of the SDHB:c.574T>C mutation, reflecting the autosomal dominant inheritance risk for paraganglioma and other associated tumors. This variant was previously reported,4 and classified as ‘pathogenic’ (rs786202732) in the online publicly available ClinVar database. This study highlights the complexity of managing such families and encourages further discussion regarding the management of asymptomatic PPGL-associated mutation carriers.
Based on biochemical secretory patterns, PPGLs can be characterized into three different phenotypes; noradrenergic (predominantly norepinephrine-secreting), adrenergic (predominantly epinephrine-secreting), and dopamine-secreting. The tumor biochemical profile depends on the localized activation of the catecholamine enzymatic machinery. The final step of converting norepinephrine to epinephrine is catalyzed by the enzyme phenylethanolamine N-methyltransferase (PNMT), enhanced by the high cortisol background in the adrenal cortex, and is predominantly restricted to normal adrenal tissue.5 This is why epinephrine is the primary hormone secreted by tumors arising from the adrenal medulla. On the other hand, PNMT levels in the paraganglia are negligible, and the primary secretion of paragangliomas (extra-adrenal tumors) is norepinephrine. This was evident in our index case, Patient II.15. The high levels of urine and serum normetanephrine in the index case and his sister, Patient II.4 were strong predictors of extra-adrenal lesions, confirmed by imaging.
The large variation in clinical presentation of PPGLs can be attributed to the hemodynamic and metabolic actions of the catecholamines produced and secreted by these tumors. In the family reported here, the 14-year-old index case, presented with severe hypertension. His mother was clinically and biochemically normal, despite having an undetected lesion in the middle mediastinum, which is the rarest location for PPGLs. His 34-year-old sister had a functioning paraganglioma with elevated normetanephrine and CgA levels, albeit being initially normotensive. However, PPGLs that are initially asymptomatic can present with symptoms during pregnancy due to (1) release of catecholamines, (2) increased intra-abdominal pressure exerted by the growing uterus, (3) fetal movements or uterine contractions, (4) abdominal palpation, (5) physical or emotional stress, (6) labor and delivery, and/or (7) spontaneous hemorrhage into the tumor.6 A possible role for estrogens has also been proposed, where they may act as growth factors, leading to tumor enlargement.7 Due to the rarity and extremely variable presentation of PPGLs, diagnosing this condition in pregnant women is a clinical challenge and a necessity to avoid complications during labor. Due to the complexity of clinical management, such patients should be catered by a multidisciplinary team (preferably an endocrinologist, obstetrician, endocrine surgeon and anesthetist) with high levels of expertise in the field. The early diagnosis of paraganglioma in Patient II.4 expedited such a management strategy during her pregnancy, which eventually resulted in favorable maternal and fetal outcomes.
Interestingly, about 14% of patients with PPGLs have no hypertension despite having chronically elevated catecholamine levels.8 The likely hypothesis for this presentation is that it may be due to adrenergic desensitization caused by adrenergic receptors undergoing sequestration, downregulation, or phosphorylation.9 Normal blood pressure is more frequent in patients discovered incidentally or in those undergoing screening for familial catecholamine-secreting tumors,10 as in the case of both the 34-year-old sister before pregnancy and the 56-year-old mother in this family. As familial PPGLs can be clinically silent, biochemical tests alone may still miss lesions, as is evident from the mother’s case. This mandates the need for radiological surveillance (RS) of asymptomatic carriers. Although the Endocrine Society clinical practice guidelines generally recommend periodic MRI-screening of asymptomatic carriers of familial PPGLs,3 there is no clear consensus in the field regarding the mode of RS or the frequency of imaging of asymptomatic carriers of hereditary PPGLs, and in particular, for those with biochemically silent PPGLs. We recommend annual MRI-screening for all carriers, starting at least 10 years earlier than the youngest age of disease manifestation in an affected family.
In pregnant women with suspected PPGLs, MRI is the first imaging choice as it provides good visualization of the abdomen, pelvis, neck, and mediastinum.11 Moreover, the sensitivity of MRI without gadolinium in pregnancy is unknown. Morever, our experience in Patient II.4’s case showed that the accurate picture was only obtained after CT with contrast as opposed to MRI without gadolinium.
Conclusion
Genetic screening has enabled the early detection of PPGLs for which early surgical intervention can significantly impact prognosis and treatment strategies to reduce morbidity and mortality. While current guidelines are clear with respect to symptomatic PPGL cases, the management of asymptomatic mutation carriers requires further elucidation. Each PPGL case should be managed in a tertiary referral center by multidisciplinary teams of specialists to provide an individualized treatment plan.
Disclosure
The authors declared no conflicts of interest.
references
- 1. Eisenhofer G, Lenders JW, Pacak K. Biochemical diagnosis of pheochromocytoma. Front Horm Res 2004;31:76-106.
- 2. Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol 2015 Feb;11(2):101-111.
- 3. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014 Jun;99(6):1915-1942.
- 4. Burnichon N, Rohmer V, Amar L, Herman P, Leboulleux S, Darrouzet V, et al; PGL.NET network. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab 2009 Aug;94(8):2817-2827.
- 5. Pillai S, Gopalan V, Smith RA, Lam AK. Updates on the genetics and the clinical impacts on phaeochromocytoma and paraganglioma in the new era. Crit Rev Oncol Hematol 2016 Apr;100:190-208.
- 6. Plu I, Sec I, Barrès D, Lecomte D. Pregnancy, cesarean, and pheochromocytoma: a case report and literature review. J Forensic Sci 2013 Jul;58(4):1075-1079.
- 7. Lenders JW. Pheochromocytoma and pregnancy: a deceptive connection. Eur J Endocrinol 2012 Feb;166(2):143-150.
- 8. Suzawa K, Yamamoto H, Ichimura K, Toyooka S, Miyoshi S. Asymptomatic but functional paraganglioma of the posterior mediastinum. Ann Thorac Surg 2014 Mar;97(3):1077-1080.
- 9. Krause M, Reinhardt D, Kruse K. Phaeochromocytoma without symptoms: desensitization of the alpha- and beta-adrenoceptors. Eur J Pediatr 1988 Feb;147(2):121-122.
- 10. Gimenez-Roqueplo AP, Caumont-Prim A, Houzard C, Hignette C, Hernigou A, Halimi P, et al; EVA Investigators. Imaging work-up for screening of paraganglioma and pheochromocytoma in SDHx mutation carriers: a multicenter prospective study from the PGL. J Clin Endocrinol Metab 2013 Jan;98(1):E162-E173.
- 11. Ahlawat SK, Jain S, Kumari S, Varma S, Sharma BK. Pheochromocytoma associated with pregnancy: case report and review of the literature. Obstet Gynecol Surv 1999 Nov;54(11):728-737.