Congenital hypothyroidism (CH) is defined as a thyroid hormone deficiency present at birth. It is one of the most common endocrine disorders in neonates as well as one of the few preventable causes of severe intellectual disability.1–3 CH is classified into permanent and transient, and in each group, there are many different etiologies. Permanent CH refers to a persistent deficiency of thyroid hormone that requires lifelong treatment. While, transient CH is a temporary deficiency of the thyroid hormone, usually discovered at birth, the infants recover to normal thyroid hormone production (euthyroidism) in the first few months or years of life.1,2,4
The majority of CH cases (80–85%) is due to thyroid dysgenesis, which includes thyroid agenesis, hypoplasia, and ectopy.1,2 Dyshormonogenesis (an error of the thyroid hormone biosynthesis) only account for 10% of cases. The remaining are due to other causes such as central hypothyroidism and thyroid hormone resistance.5–12
Many known causes are attributed to the transient form of CH such as a maternal and neonatal iodine deficiency or excess, maternal antithyroid drugs, transplacental passage of the maternal thyroid-stimulating hormone (TSH) receptor-
blocking antibodies.2,9
Before the onset of newborn screening programs, the incidence of CH was on the range of 1:7000–10 000 and based on clinical manifestations. Screening for CH was introduced in 1974 and has become common practice.10,11 After starting the newborn screening program, the incidence was initially reported to be in the range of 1:3000 to 1:4000. With more experience from regional and national screening programs, it has become apparent that the incidence varies by geographic location.1,10 In the USA, the incidence was 1:2372 live births, in Europe it was 1:3000 live births, and in Japan 1:5700 live births.3,13 On the other hand, the incidence was high in areas with high consanguinity rates like in Saudi Arabia (1:2500 live births), Jordan (1:2242 live births), and Iran (1:1000 live births).14–18 In Oman, we could not find any data to compare with it. In terms of sex, studies found CH is more common in females with the female-to-male ratio of 3:1.3.17
With increased sensitivity and accuracy of TSH methods, the USA and other programs around the world have switched from a primary thyroxine (T4), follow-up TSH approach to a primary TSH test. Also, lowering TSH cutoff allowed more infants with the milder form of CH to be detected earlier.4,11
The majority of neonates with CH are asymptomatic at birth, which will result in severe neurodevelopmental impairment if treatment is delayed.1–3 Therefore, it is mandatory to screen neonates for CH for early diagnosis and treatment.1 Many theories tried to explain the possible causes for delayed clinical manifestations of CH, and the most popular ones are attributed to the placental passage of maternal thyroid hormone. Also, the most common forms of CH have some moderately functioning thyroid tissue.9,19 Approximately 10% of infants with CH have associated congenital anomalies with the cardiac defects being the most common anomalies, but other anomalies of the nervous system and eyes have been reported.20
In 2004, the national screening program for CH was started in Oman for all newborns.21 This study aimed to determine the demographic features and the etiology of the CH in Oman.
Methods
This retrospective study was conducted at the National Diabetes and Endocrine Center in Oman. The patients treated in this center are referred from all governorates in the country and are representative of the population as a whole. Ethical approval was obtained by the scientific research committee of the Royal Hospital, Muscat, Oman (SRC #82/2017).
Data were collected from the patients’ records from January 2004 to December 2016 (a total of 13 years).
In this research, the inclusion criteria were all Omani children newly diagnosed with CH from January 2004 to December 2016 that were followed-up at the National Diabetes and Endocrine Center at the Royal Hospital. Exclusion criteria were children with transient hypothyroidism. The data collected from the patients’ records includes:
- Demographic data such as current age, sex, gestational age, type of delivery, birth anthropometric (weight, height, and head circumference), and governorate.
- Clinical manifestations of CH, which include feeding problems, failure to thrive, constipation, prolonged jaundice, coarse face, macroglossia, goiter, mottled skin, or hypothermia, and other possible congenital anomalies.
- Lab investigations (cord TSH, pre-treatment TSH and T4, and day of investigation).
- Date of starting T4.
- Thyroid scan reports.
Developmental assessments were done for each patient on the first presentation and also were repeated on each follow-up visit. During the data collection process, if the patient was found to have developmental delay, then an extensive review was done for the patient medical file to exclude the presence of the secondary causes of the developmental delay apart from the CH such as trisomies or other medical causes.
A cutoff level for the cord TSH remains controversial worldwide. For example, in Turkey, the cutoff level is > 15 mIU/L, in Jordan it is > 20 mIU/L.17,18 In Oman, according to the guidelines of the Ministry of Health, the cutoff levels for cord TSH is > 40 mIU/L, which might increase the possibility of false-negative results.22 Newborns with positive results (TSH > 40 mIU/L) were called for a further assessment for symptoms and signs of CH, and then venous blood samples were collected for free T4 and TSH tests. The results were available within 1–5 days.
Regarding thyroid scan for the confirmed cases with CH, Tc-99m pertechnetate thyroid scintigraphy was done to determine the underlying etiology before starting T4 therapy, especially for those born at the Royal Hospital due to the availability of the scan in this hospital. For newborns referred from peripheral hospitals, thyroid scintigraphy was postponed until the age of 3 years for the benefit of starting T4 earlier. Thyroid ultrasonography was performed when the scintigraphy did not show any uptake in the thyroid gland area to confirm the diagnosis of thyroid agenesis. Infants diagnosed with CH were followed closely for the first three years of life to distinguish between the permanent and the transient CH.
Information was initially entered into data collection sheets and then transferred to the Epidata version 3.1, which was then analyzed using SPSS Statistics (IBM Corp. Released 2016. IBM SPSS Statistics for Windows, Version 24.0. Armonk, NY: IBM Corp.). Descriptive statistics, such as mean, median, standard deviation, frequency, and percentages, were used to describe the demographic and the clinical parameters. Fisher’s exact test was performed to find the association between treatment delay and developmental delay. A crude odds ratio was also reported for the association between the treatment delay and the developmental delay. A p-value < 0.050 was considered statistically significant.
Results
A total of 104 patients were newly diagnosed with the CH at the National Diabetes and Endocrine Center between January 2004 and December 2016. However, only 96 patients (92.3%) were included in this study, and eight patients (7.7%) were excluded due to different reasons (five patients had transient hypothyroidism and three patients were expatriates).
Out of the 96 patients with CH, 43 (44.8%) were males, and 53 (55.2%) were females giving a female-to-male ratio of 1.2:1. The average age was 6.9 years. Regional variation in the incidence was observed between governorates. The most reported cases were from Muscat with 48 (50.0%) patients, followed by Al Batinah (n = 10, 10.4%). Cases from the other governorates were as follows: 14 (14.6%) patients were from A’Sharqiyah, 12 (12.5%) from A’Dhahirah, and eight (8.3%) from A’Dakhiliyah. Only two (2.1%) patients were from both Musandam and Al Wusta governorates. No patients were from the Dhofar or Al Buraymi governorates.
The majority of the children were born by spontaneous vaginal delivery (n = 66, 68.8%). The average birth weight was 2.9 kg. The average birth height and head circumferences were 51.5 cm and 33.6 cm, respectively. The average gestational age was 38.3 weeks. The average pre-treatment TSH level was 104.5 mIU/L. The median number of days for repeated investigation and treatment initiation was three days. Other details are given in Table 1.
Table 1: Clinical characteristics of the patients.
|
Delivery type |
|
|
SVD |
66 (68.8) |
|
Elective LSCS |
1 (1.0) |
|
Emergency LSCS |
13 (13.5) |
|
Unknown |
16 (16.7) |
|
Birth weight, mean ± SD, kg |
2.9 ± 0.6 |
|
Birth height, mean ± SD, cm |
51.5 ± 3.5 |
|
Head circumference at birth, mean ± SD, cm |
33.6 ± 2.2 |
|
Gestational age, mean ± SD, weeks |
38.3 ± 2.5 |
|
Cord TSH, mean ± SD, mIU/L |
92.2 ± 37.9 |
|
Pre-treatment TSH, mean ± SD, mIU/L |
104.5 ± 74.1 |
|
Pre-treatment T4, mean ± SD, pmol/L |
10.3 ± 6.1 |
|
Day of investigation, median (min-max) |
3 (1–240) |
SVD: spontaneous vaginal delivery; LSCS: lower segment cesarean section, TSH: thyroid-stimulating hormone; T4: thyroxine.
Table 2: Clinical manifestations of congenital hypothyroidism.
|
Asymptomatic |
84 (87.5) |
|
Constipation |
4 (4.2) |
|
Feeding problems |
3 (3.1) |
|
Prolonged jaundice |
2 (2.1) |
|
Coarse face |
1 (1.0) |
|
Macroglossia |
1 (1.0) |
|
Goiter |
1 (1.0) |
The majority of patients were asymptotic (n = 84; 87.5%). Constipation was the most reported symptom (n = 4; 4.2%) [Table 2]. Regarding the associated congenital anomalies, it was found that only 18 patients (18.8%) in which the cardiac defects were the most common.
Regarding the etiology of CH, a thyroid scan was done in 78 (81.3%) patients. While the remaining 18 patients (18.8%) thyroid scan was not done due to different reasons. For the majority of the patients, the scan was not performed due to age restriction, as it is recommended that T4 therapy is continued until the age of three to protect brain development. Therefore, thyroid scans were planned to be done once they were three years of age so that it would be safe to discontinue T4 one month before the scan.
Out of the 78 patients, thyroid dysgenesis (ectopic and agenesis) was the most common etiology with a total of 40 cases (51.3%) followed by dyshormonogenesis with 35 (44.9%) cases. Only three (3.8%) patients had CH due to central hypothyroidism [Figure 1].
Out of the 96 patients, we were able to find the initiation day of the T4 therapy for 83 patients (86.5%), while for the remaining 13 patients (13.5%) there was no mention regarding the starting date due to missing information in the referral letters from the peripheral hospitals. Out of these 83 patients, the majority (n = 72, 86.7%) were started on the T4 within 30 days of life with an average age of treatment commencement of three days. For the remaining 11 patients (13.3%), there was a delay for more than 30 days in starting the treatment due to compliance issues [Figure 2].
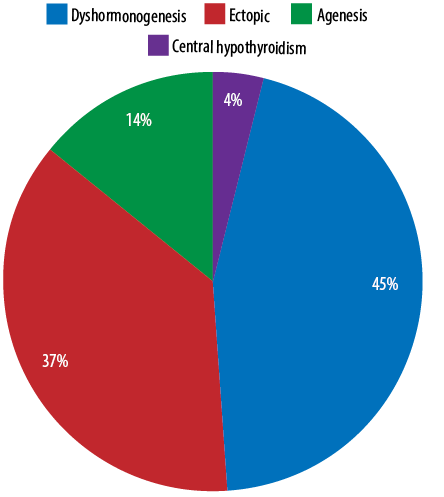
Figure 1: Etiology of congenital hypothyroidism
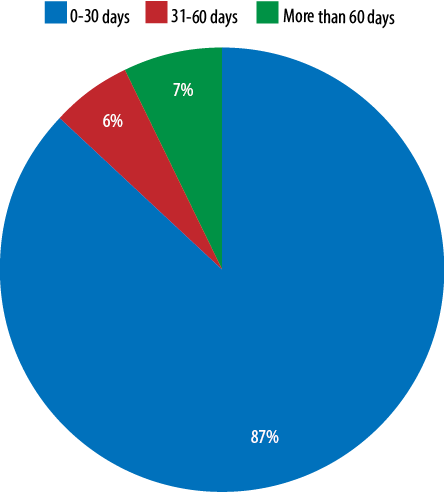
Figure 2: Initiation day of thyroxine therapy.
The patients in this study were divided into two groups (delay and no delay) based on the delay in starting T4 therapy. Starting the treatment after 30 days was considered as treatment delay. Also, patients were divided into two groups based on the developmental delay (delay and no delay). We aimed to look for any association between the developmental delay and the delay in starting the T4 therapy among these patients. Fisher’s exact test showed a statistically significant association between the two (p < 0.001). Out of the 11 patients who had a delay in starting the T4 therapy, eight (72.7%) had a developmental delay, and three (27.3%) had no developmental delay [Table 3]. The crude (unadjusted) odds ratio for the association between the treatment delay and the developmental delay was 16.53 (95% CI: 3.74–73.03, p < 0. 001). This finding further emphasized that there is a statistically significant increased risk (16.5 times) for developmental delay in children with CH who are not identified and treated with T4 therapy in a timely manner.
Reporting the overall incidence of CH in Oman was not possible due to the type of study, which involves only one center. Unfortunately, there is no available public national data regarding the incidence of positive cord TSH result. After contacting the District Health Information System at the Ministry of Health in Oman, they were only able to provide the statistics from 2012 to 2016 with many missing data. Therefore, the incidence of CH in Oman was difficult to estimate.
Table 3: Association between the delay in starting the thyroxine therapy and the developmental delay.
*Statistically significant, Test: ’Fisher’s exact test.
Discussion
CH is the most common thyroid disorder, with an estimated population prevalence in the childhood of 0.14%. Thyroid hormone plays a major role in the functions of various organs, especially in brain development.2,23 Fetal thyroid predominantly produces ‘reverse T3’, which is largely an inactive hormone, and this is why a fetus depends on their mothers to provide enough T4 hormone for their needs. After birth, there is a surge in the level of the TSH, which is accompanied by a marked rise in T4 and T3 levels. Then, TSH declines to the normal adult range within a week.9
The concept of neonatal screening started in 1963 for phenylketonuria (PKU). Ten years later, this concept was extended to the CH. CH is seven-fold more frequent than PKU, and the early treatment of CH leads to a more drastic effect on the neuropsychological outcome than the dietary restriction for PKU. Also, the cost of CH screening is not comparable to the cost of long-term care of patients with the physical and mental disability, and this why most countries applied it as part of their national screening program. In Oman, the national screening for CH started in 2004,21 and this why we choose to include patients from 2004.
Most of the newborns are asymptomatic at birth, and only 5–10% of affected newborns have clinical signs or symptoms of CH. Infants with the severe CH lasting more than 4-6 weeks may present with the symptoms and signs of CH such as poor feeding, constipation, lethargy or excessive sleeping, failure to thrive, large anterior fontanelles, dry skin, prolonged jaundice, mottling, umbilical hernia, macroglossia, or coarse facial feature.2,24 In our study, most of the patients were asymptotic (n = 84, 87.5%), which is considered slightly low in comparison to the worldwide prevalence (90–95% are asymptotic). The most obvious explanation for this low prevalence is due to the delay in starting the T4 therapy beyond one month in those patients (n = 11, 13.3%).
Failure to a recognition of the CH in the neonatal period will lead to irreversible, permanent central nervous system damage, which will result in physical and mental disability. Early treatment of infants diagnosed with CH was associated with a normal mean IQ. Preferably, the diagnosis of the CH must be confirmed within the first 14 days, and treatment should be started immediately (ideally within the first month of life).1,25 In this study, most newborns diagnosed with CH were started on treatment before the age of one month (n = 72, 86.7%), with an average age of starting the treatment of three days. Because of that, this group of patients had healthy development. For the remaining 11 patients (13.3%), there was a delay in starting treatment, which led to a developmental delays in this group compared to those who had treatment earlier.
The majority of CH cases (80–85%) are due to thyroid dysgenesis, while thyroid dyshormonogenesis accounts for only 10%.5,6 But in this study, dyshormonogenesis accounted for 44.9%, which could be explained by the high consanguinity rate in Oman, as dyshormonogenesis tends to be inherited in an autosomal recessive pattern. However, due to the limitations of the medical records regarding the missing information about consanguinity, we could not prove it statistically.
The American Academy of Pediatrics recommended an initial starting dose of T4 of 10–15 µg/kg/day. Then, thyroid function tests should be monitored every 1–2 months in the first six months of life with the goal of the free T4 level in the upper half of normal range and the TSH in the normal range. If permanent hypothyroidism has not been diagnosed before the onset of therapy, it is recommended that treatment is discontinued sometime after the age of three years for 30 days to determine permanency of the hypothyroidism.1–3
The first major limitation of this study concerns the medical records for missing information about consanguinity, birth history, date of starting the T4, especially for the cases referred from a peripheral hospital.
Conclusion
This is the first epidemiological study conducted in Oman that highlights the unique demographic and the etiology features of CH. Dyshormonogenesis has a high prevalence in the Omani population compared to other nations (i.e., European and North American origin). However, more studies are needed to focus on possible risk factors such as consanguinity. The neurological sequelae in our patients was higher compared to the worldwide prevalence, which was mainly due to the delay in starting the T4 therapy due to compliance issues, which emphasized the need for extensive counseling about the sequelae of the disease.
Disclosure
The authors declare no conflict of interest. No funding was received for this study.
Acknowledgements
We acknowledge all hospital staff who contributed to this project.
references
- 1. LaFranchi SH, Huang SA. Thyroid development, and physiology. In: Kliegman R, Nelson WE, editors. Nelson textbook of pediatrics. 20th ed. Philadelphia: Elsevier; 2016. p. 2663-2675.
- 2. Rastogi MV, LaFranchi SH. Congenital hypothyroidism. Orphanet J Rare Dis 2010 Jun;5:17.
- 3. Olney RS, Grosse SD, Vogt RF Jr. Prevalence of congenital hypothyroidism–current trends and future directions: workshop summary. Pediatrics 2010 May;125(Suppl 2):S31-S36.
- 4. Harris KB, Pass KA. Increase in congenital hypothyroidism in New York State and in the United States. Mol Genet Metab 2007 Jul;91(3):268-277.
- 5. Stoppa-Vaucher S, Van Vliet G, Deladoëy J. Variation by ethnicity in the prevalence of congenital hypothyroidism due to thyroid dysgenesis. Thyroid 2011 Jan;21(1):13-18.
- 6. van Tijn DA, de Vijlder JJ, Verbeeten B Jr, Verkerk PH, Vulsma T. Neonatal detection of congenital hypothyroidism of central origin. J Clin Endocrinol Metab 2005 Jun;90(6):3350-3359.
- 7. Kopp P. Pendred’s syndrome and genetic defects in thyroid hormone synthesis. Rev Endocr Metab Disord 2000 Jan;1(1-2):109-121.
- 8. Karamizadeh Z, Dalili S, Sanei-Far H, Karamifard H, Mohammadi H, Amirhakimi G. Does congenital hypothyroidism have different etiologies in iran? Iran J Pediatr 2011 Jun;21(2):188-192.
- 9. Herbstman J, Apelberg BJ, Witter FR, Panny S, Goldman LR. Maternal, infant, and delivery factors associated with neonatal thyroid hormone status. Thyroid 2008 Jan;18(1):67-76.
- 10. Kaye CI, Accurso F, La Franchi S, Lane PA, Northrup H, Pang S, et al; Committee on Genetics. Introduction to the newborn screening fact sheets. Pediatrics 2006 Sep;118(3):1304-1312.
- 11. Fisher DA. Effectiveness of newborn screening programs for congenital hypothyroidism: prevalence of missed cases. Pediatr Clin North Am 1987 Aug;34(4):881-890.
- 12. Straussman S, Levitsky L. Congenital hypothyroidism –monitoring thyroid function in infants. Eur Endocrinol 2012;8:53-56 .
- 13. Gu YH, Kato T, Harada S, Inomata H, Saito T, Aoki K. Seasonality in the incidence of congenital hypothyroidism in Japan: gender-specific patterns and correlation with temperature. Thyroid 2007 Sep;17(9):869-874.
- 14. Abu-Osba YK, Mallouh A, Salamah M, Hann R, Thalji A, Hamdan J, et al. Comprehensive newborn screening program: ARAMCO experience, the national need and recommendations. Ann Saudi Med 1992 May;12(3):235-240.
- 15. Bacchus R, Williams S, Joyce B, Sabagh TO, Khan M, Paterson W. Neonatal screening for congenital hypothyroidism in Riyadh. Saudi Med J 1988;9(6):588-595.
- 16. Al Jurayyan N, Al Jurayyan R. Congenital hypothyroidism and neonatal screening in Saudi Arabia. Curr Pediatr Res 2011;16(1):31-36.
- 17. Dilli D, Çzbaş S, Acıcan D, Yamak N, Ertek M, Dilmen U. Establishment and development of a national newborn screening programme for congenital hypothyroidism in Turkey. J Clin Res Pediatr Endocrinol 2013;5(2):73-79.
- 18. Alawneh H. Incidence of congenital hypothyroidism in Jordan. Menoufia Med J 2014;27(2):503-506 .
- 19. American Academy of Pediatrics. Newborn screening for congenital hypothyroidism: recommended guidelines. Pediatrics 1993 Jun;91(6):1203-1209.
- 20. Abo El-Magd M, El-Maraghy M, Abdelrahim M, El-Meguid K, Meabed M. Prevalence of congenital heart diseases in children with congenital hypothyroidism. Arch Clin Exp Surg 2013;2(2):85-91 .
- 21. Reduce mortality rate among children younger than 5 years and neonate in Oman. 2015 [cited 2019 May]. Available from: http://alwatan.com/details/80907.
- 22. Ministry of Health. Congenital hypothyroidism guidelines for neonatal screening and management. June 2010 ed. Oman: 2010. [cited 2019 May]. Available from: https://www.moh.gov.om/documents/272928/4017900/CONGENITAL+HYPOHYROIDISM-1.pdf/7750d759-3f49-77f8-088f-8196f2c6da5f.
- 23. Rezaeian S, Poorolajal J, Moghimbegi A, Esmailnasab N. Risk factors of congenital hypothyroidism using propensity score: a matched case-control study. J Res Health Sci 2013 Sep;13(2):151-156.
- 24. Deladoëy J, Ruel J, Giguère Y, Van Vliet G. Is the incidence of congenital hypothyroidism really increasing? A 20-year retrospective population-based study in Québec. J Clin Endocrinol Metab 2011 Aug;96(8):2422-2429.
- 25. el-Desouki M, al-Jurayyan N, al-Nuaim A, al-Herbish A, Abo-Bakr A, al-Mazrou Y, et al. Thyroid scintigraphy and perchlorate discharge test in the diagnosis of congenital hypothyroidism. Eur J Nucl Med 1995 Sep;22(9):1005-1008.