Hypoglycemia is a serious complaint in children that needs a detailed history, physical examination, and multiple diagnostic tests to confirm its etiology. The differential diagnosis of recurrent hypoglycemia in children includes endocrine and metabolic disorders.1 Adrenal insufficiency, hyperinsulinism, and hypopituitarism are the leading endocrine causes of recurrent hypoglycemia in the pediatric population. The metabolic causes include glycogen storage disease, gluconeogenesis disorders, fatty acid oxidation defects, ketone synthesis or utilization defects, succinyl-CoA transferase deficiency, mitochondrial respiratory chain defect, and idiopathic ketotic hypoglycemia.1
Congenital hyperinsulinism (CHI) due to beta-cell hyperplasia is a well-known clinically and genetically heterogeneous entity causing severe hypoglycemia in neonates and infants.2 During childhood and adolescence, insulin-secreting neuroendocrine tumors (insulinoma) are rare findings. Only a few cases of insulinoma have been reported in children worldwide,2–6 and there are very few case reports from Saudi Arabia, the youngest was a 16-year-old girl.7,8 The clinical presentations may vary but mostly include neuroglycopenic symptoms due to severe hypoglycemia. Most affected children were mislabeled as having a neurological disease and had extensive neurological assessments before reaching the correct diagnosis, sometimes with a delay of several months or years. In addition, localization of insulinoma may prove to be difficult.6 We reviewed the current literature with a focus on the recommended diagnostic and therapeutic strategies in adults as only a few references in children are available. Here we report a successful diagnosis and treatment of a Saudi child who presented with a persistent hyperinsulinemic state.
Case report
A seven-year-old boy with a history of frequent hypoglycemia episodes (over one year) was referred to our tertiary care center for a diagnostic opinion. The patient was maintained on small doses of diazoxide (3 mg/kg/day) from the referring hospital.
The patient was admitted for a diagnostic induced hypoglycemia test. After eight hours of fasting, he developed hypoglycemia (glucose level 1.9 mmol/L), and a critical sample showed his insulin level was 11.7 µU/mL (normal level 6–27 µU/mL) equivalent to 70 pmol/L. His C-peptide level was found to be 533 pmol/L (normal level 364–1655 pmol/L). Other elements of the samples (ketones, cortisol, adrenocorticotropic hormone, growth hormone, lactic acid, ammonia, free fatty acid level, serum acetylcarnitine profile, and urine organic acid screening)8 were all normal. The blood glucose (BG) response to glucagon was 5 mmol/L. However, his hypoglycemia persisted and required dextrose 10% bolus and infusion in addition to diazoxide and octreotide. We were able to wean him from intravenous (IV) fluids after 24 hours. Magnetic resonance imaging (MRI) of the abdomen showed a normal pancreas and liver, as reported alongside the computed tomography (CT) images taken by the referring hospital reported as normal. DNA sample was extracted to determine any genetic causes of hyperinsulinism.
During follow-up over months in the clinic, the patient started to require increasing doses of diazoxide 75 mg orally three times daily (10 mg/kg/day) and octreotide 150 mcg subcutaneous twice daily (15 mcg/kg/day).
One day, the patient missed the evening dose of octreotide, and the next morning he developed severe hypoglycemia symptoms and was disoriented. A home glucose test was 1 mmol/L (20 mg/dL), and he rapidly lost consciousness. He received 1 mg glucagon and was brought to our emergency department. He was unconscious and severely hypoglycemic (1.9 mmol/L).
Patient's hypoglycemia was very resistant to multiple glucose D10% boluses (5 mL/kg) and infusion, three doses of glucagon 1 mg, two doses of octreotide 150 mcg, two doses of hydrocortisone 50 mg/m2/dose, two doses of diazoxide 125 mg, and glucose D12.5% infusion (15 mg/kg/min). With all these measures, his maximum glucose level was 4–6 mmol/L, and it was fluctuating rapidly down to 1 mmol/L. His level of consciousness only showed some improvement when his glucose level was > 2 mmol/L.
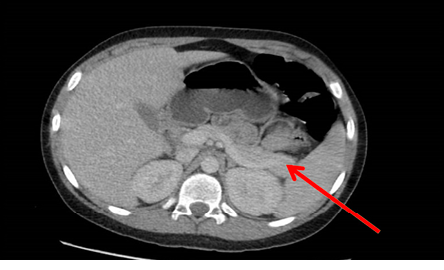
Figure 1: CT scan of the adomen revealed a pancreatic tail tumor.
Table 1: Genetics testing summary of a seven-year-old boy with frequent hypoglycemia.
|
Molecular genetic analysis of known familial hyperinsulinism genes by next-generation sequencing |
ABCC8, KCNJ11, GCK, GLUD1, HADH, HNF4A, SLC16A1, and UCP2 |
Negative |
We observed the patient in the pediatric intensive care unit. In the next 24 hours, he needed maximum doses of diazoxide treatment (15 mg/kg/day six hourly) and octreotide treatment (40 mcg/kg/day four hourly) with tapering of D12.5% infusion. IV fluids were discontinued when glucose level maintained > 4 mmol/L.
Investigations during this severe hypoglycemia revealed BG was 1.9 mmol/L, and a very high insulin level of 20 µU/mL (105 pmol/L).
Abdominal CT scan was repeated with frequent sequencing to assess the pancreas, which revealed a 2.5 × 1 cm mass at the tail of the pancreas, suggesting an insulinoma [Figure 1].
The patient underwent laparoscopic enucleation to remove the tumor from the pancreatic tail. The patient was observed post-operatively in the pediatric intensive care unit for 24 hours and was then shifted to the ward. During the post-operative time, he was off medication and IV fluids, tolerating oral intake, and BG were within normal levels (6–8 mmol/L). He was discharged home in good condition four days post-surgery.
Specimen histopathology report confirmed the diagnosis of pancreatic tail insulinoma well-differentiated neuroendocrine tumor.
Outpatient follow-up for the last 12 months showed a well-controlled blood sugar level with no hypoglycemia and an improvement in school performance and lifestyle with the disappearance of hirsutism that occurred as a side effect of diazoxide use.
There was no significant family history of similar illness. All molecular genetic analysis of known familial hyperinsulinism genes (ABCC8, KCNJ11, GCK, GLUD1, HADH, HNF4A, SLC16A1, and UCP2) by next-generation sequencing showed no mutation. In addition, MEN1 mutation screening was negative [Table 1].
Discussion
CHI is the most common cause of hypoglycemic hyperinsulinemia in infants usually caused by genetic defects, though it can be at any age.1 In contrast, insulinoma is the most prevalent form of endogenous hypoglycemic hyperinsulinemia in adults, but only in 20% of cases is the correct diagnosis was established within one year of symptoms onset.9,10 To date, < 30 cases of insulinoma at a young age have been published, and the rarity makes a timely and accurate diagnosis even more difficult.11 A diagnostic fasting test was done for 96 Omani children aged eight days to 10 years in a tertiary hospital in Oman for hypoglycemia investigation; 14 (15%) were confirmed to have hyperinsulinism. None of those cases confirmed to be due to insulinoma, suggesting it is a rare condition in Gulf countries.1 Among the Saudi population, there are few cases in adults; the youngest reported case was a 16-year-old girl.7,8 Our patient was only seven years old at presentation. Initially, our patient was evaluated for a seizure disorder, which is not an uncommon misleading workup for insulinoma,12 but not all neuroglycopenic symptoms, namely, deterioration of school performance and behavior, could be explained by focal epilepsy. Given low plasma glucose (≤ 2.5 mmol/L) in a patient appearing healthy and alleviation of neuroglycopenic symptoms after intake of carbohydrates, denoted as the Whipple triad, hypoglycemic hyperinsulinism may be assumed as the underlying cause, namely if elevated levels of insulin and C-peptide are measured, as in our case. Serum insulin levels should normally be low at the times of hypoglycemia to below the limits of detection of the assay. Thus, any detectable level of insulin at the critical time of hypoglycemia can be considered abnormal and must be viewed as evidence of inappropriate insulin secretion.13
Evaluation of the glucose response to glucagon injection at the end of the fast or during a spontaneous hypoglycemia episode provides a quick and reliable measure of increased insulin effect and helps establish the diagnosis of hyperinsulinemia. An increase in serum glucose level of > 1.7 mmol/L (30 mg/dL) in response to glucagon administration (1 mg IV or intramuscular) suggests inappropriate preservation of liver glycogen at the time of hypoglycemia and indicates suppression of liver glycogenolysis by excess insulin effect.13
Insulinoma is poorly managed medically, and surgical options are always indicated.14 It often presents a diagnostic challenge both clinically and radiologically, and even in the hands of an expert radiologist, the image could be reported as negative.15 Progressive resistance for anti-insulin medications and increase frequency of hypoglycemic attacks should put the diagnosis of insulinoma top of the list even with no radiological clues.
The primary role of imaging is to localize the lesion preoperatively. The major challenge for the detection of insulinoma has been the small size of the lesion. Using modern imaging techniques, such as 18-fluorine-DOPA positron emission tomography, is useful for patients with insulinoma, especially with negative CT and MRI, and can hold unnecessary investigations.16 Unfortunately, it was not available at our institute.
Conclusion
We reported here the youngest insulinoma case described so far in Saudi Arabia. Our case emphasizes the importance of putting insulinoma at the top of the differential diagnosis list in any child after infancy who present with hypoglycemic hyperinsulinism, with a careful review of imaging studies and monitoring the response for anti-insulin medications and the frequency of hypoglycemic attacks. Accurate diagnosis and clear localizing of the tumor can save the patient from unnecessary total pancreatectomy and post-surgical consequences.
Disclosure
The authors declared no conflict of interest.
Acknowledgements
We extend our thanks and appreciation to Radiology, Surgery, and Laboratory Departments at National Guard Medical City, Jeddah, Saudi Arabia, who provided help and advice to us.
references
- 1. Bappal B, Mula-Abed WA. Evaluation of diagnostic fasting in the investigation of hypoglycemia in children omani experienc. Oman Med J 2007 Oct;22(3):36-41.
- 2. Arnoux JB, de Lonlay P, Ribeiro MJ, Hussain K, Blankenstein O, Mohnike K, et al. Congenital hyperinsulinism. Early Hum Dev 2010 May;86(5):287-294.
- 3. Grosfeld JL, Vane DW, Rescorla FJ, McGuire W, West KW. Pancreatic tumors in childhood: analysis of 13 cases. J Pediatr Surg 1990 Oct;25(10):1057-1062.
- 4. Jaksic T, Yaman M, Thorner P, Wesson DK, Filler RM, Shandling B. A 20-year review of pediatric pancreatic tumors. J Pediatr Surg 1992 Oct;27(10):1315-1317.
- 5. Karachaliou F, Vlachopapadopoulou E, Kaldrymidis P, Simatos G, Zacharea M, Spanidou-Karvouni E, et al. Malignant insulinoma in childhood. J Pediatr Endocrinol Metab 2006 May;19(5):757-760.
- 6. Gozzi Graf T, Brändle M, Clerici T, l’Allemand D. Insulinoma: only in adults?-case reports and literature review. Eur J Pediatr 2014 May;173(5):567-574.
- 7. Arafah M, Kfoury H, Naseem S. Hypoglycemia, a rare presentation of a solid pseudopapillary neoplasm of the pancreas: a case report. The Turkish journal of gastroenterology: the official journal of Turkish Society of Gastroenterology 2011;22:544-547.
- 8. Fouda MA, Malabu UH. Insulinoma in Saudi Arabia: a twenty-year hospital study. Med J Malaysia 2008 Jun;63(2):182-184.
- 9. Chen X, Cai WY, Yang WP, Li HW. Pancreatic insulinomas: diagnosis and surgical treatment of 74 patients. Hepatobiliary Pancreat Dis Int 2002 Aug;1(3):458-461.
- 10. Mohnike K, Blankenstein O, Christesen HT, De Lonlay J, Hussain K, Koopmans KP, et al. Proposal for a standardized protocol for 18F-DOPA-PET (PET/CT) in congenital hyperinsulinism. Horm Res 2006;66(1):40-42.
- 11. Ginsberg-Fellner F, Rayfield EJ. Metabolic studies in a child with a pancreatic insulinoma. Am J Dis Child 1980 Jan;134(1):64-67.
- 12. Kao KT, Simm PJ, Brown J. Childhood insulinoma masquerading as seizure disorder. J Paediatr Child Health 2014 Apr;50(4):319-322.
- 13. De León DD, Stanley CA. Determination of insulin for the diagnosis of hyperinsulinemic hypoglycemia. Best Pract Res Clin Endocrinol Metab 2013 Dec;27(6):763-769.
- 14. Peranteau WH, Palladino AA, Bhatti TR, Becker SA, States LJ, Stanley CA, et al. The surgical management of insulinomas in children. J Pediatr Surg 2013 Dec;48(12):2517-2524.
- 15. Das CJ, Debnath J, Gupta AK, Das AK. MR imaging appearance of insulinoma in an infant. Pediatr Radiol 2007 Jun;37(6):581-583.
- 16. Imperiale A, Sebag F, Vix M, Castinetti F, Kessler L, Moreau F, et al. 18F-FDOPA PET/CT imaging of insulinoma revisited. Eur J Nucl Med Mol Imaging 2015 Mar;42(3):409-418.