Novel copy number variations (CNVs) are considered as an important reason for numerous neurodevelopmental disorders including intellectual disability (ID). CNVs are operationally defined as genomic deletions or amplifications greater than 1 kb in size. Typically, CNVs involve regions containing several to dozens of genes, often with multiple candidates in the smallest region of overlap (SRO) between similar cases. Thus, the likely candidate genes within the SRO can be proposed as the main drivers of the phenotype.
To date, four males with an Xp11.22 deletion reported in three families have been described to cause a new chromosomal X-linked syndrome.1 The Xp11.22 region comprises approximately 5 Mb of DNA (chrX:49,800,001–54,800,000, hg19). It is a gene-rich region within a critical interval for several neurodevelopmental disorders.2,3 The four patients reported had a small overlapping deletion spanning ~ 430 kb in Xp11.22 and included our candidate genes: CENPVL1, CENPVL2, G1 To S Phase Transition 2 (GSPT2), and melanoma antigen (MAGE) Family Member D1 (MAGED1).
All four patients had ID, developmental delay, hypotonia, joint hypermobility, and relative macrocephaly [Table 1]. Loss of GSPT2 and MAGED1 gene function was proposed to contribute to the ID and developmental delay seen in these deletions.1 Herein, we describe a detailed clinical and molecular description of a patient with the smallest reported deletion (~ 334 kb) within Xp11.22.
Table 1: Summary of the phenotype of patients reported with Xp11.22 deletion syndrome.
|
Xp11.22 deletion
minimum (hg19) |
chrX:50,847,688–51,773,705 |
chrX:51,079,343–
51,912,188 |
chrX:51,357,052–
52,838,176 |
chrX:51,357,052–
52,838,176 |
chrX:51,442,546-51,776,830 |
|
Xp11.22 deletion
maximum (hg19) |
chrX:50,789,912–51,786,912 |
chrX:51,079,341–
51,990,483 |
ND |
ND |
ND |
|
Age, years |
3 years 8 months |
7 years 9 months |
6 |
4 |
5 |
|
Gender |
Male |
Male |
Male |
Male |
Male |
|
Intellectual disability/
developmental delay |
+ |
+ |
+ |
+ |
+ |
|
Hypotonia |
+ |
+ |
+ |
ND |
+ |
|
Joint hypermobility |
+ |
+ |
+ |
+ |
+ |
|
Relative
macrocephaly |
+ |
+ |
+ |
+ |
+ |
|
Other growth
problems |
None |
Failure to thrive |
Failure to thrive, short stature |
Short stature |
Failure to thrive, short stature |
|
Other medical
problems |
Congenital muscular torticollis, laryngomalacia,
right-sided cryptorchidism, right-sided inguinal
hernia, gastroesophageal reflux disease, food
allergies. |
Gastroesophageal reflux
disease. |
Hypermetropia,
intermittent exotropia,
arthralgias. |
Exotropia, amblyopia. |
Cleft palate. |
ND: not determined.
Case report
The male proband was born after uneventful term pregnancy; his birth weight was 2.5 kg, head circumference was 34.4 cm, and length was 56 cm. He did not require resuscitation and had normal APGAR scores. Postnatally, he was noted to have a cleft palate, which was repaired at the age of one. The proband was the first child of healthy consanguineous parents. The remainder of the family history is noncontributory.
He had a delay in acquiring developmental milestones; he sat at the age of seven months, walked at 18 months, was able to go upstairs using alternate feet at the age of three years, and ran at four years old. At the age of five, he was able to hold a pencil, scribble, draw a circle, and copy letters. At the age of five, he had only 20 words, could not join two words, and understood simple commands only. He was able to identify two colors and count from one to three. He was toilet trained at the age of three and a half years. Detailed psychoeducational assessment was completed at the age of five and a half years using the Stanford–Binet Intelligence Scale Fifth Edition (SB5). The test scores were as follows: full-scale IQ 56 (mildly impaired or delayed), nonverbal IQ 66 (mildly impaired or delayed), and verbal IQ 47 (moderately impaired or delayed).
Growth parameters determined at five years old were as follows: height 104.5 cm (tenth percentile), weight 14.7 kg (third percentile), and head circumference 51.5 cm (fiftieth percentile). Facial features included a prominent forehead and bilateral low set ears with overfolded helices. His skeletal examination showed joint hypermobility. Otherwise, his general and neurological exams were normal. At the age of five years and six months, his brain magnetic resonance imaging was reported as normal.
The DNA sample was sent to Sistemas Genómicos, Spain. The chromosomal microarray was completed using 4x180 CGH+SNP Microarray Kit (SurePrint G3 Human, Agilent Technologies). The array’s results were analyzed with Agilent ® CytoGenomics v. 4.0.3.12 software, and the Aberration Detection Method 2 (ADM-2: 6.0) algorithm was used to identify chromosomal aberrations. The chromosomal microarray showed a 334 kb deletion at Xp11.22 [Figure 1] with genomic coordinates chrX:51,442,546-51,776,830, encompassing two pseudogenes CENPVL1, CENPVL2 and two protein-coding genes, MAGED1 and GSPT2. He inherited this deletion from his asymptomatic mother.
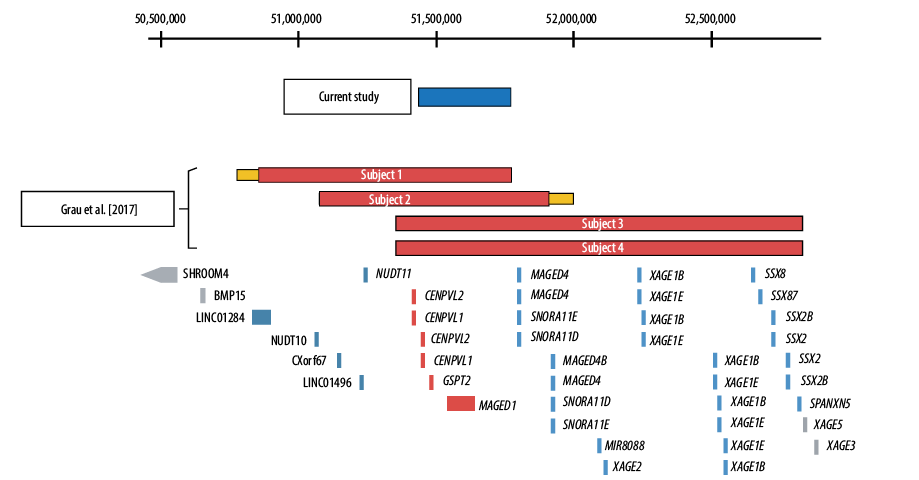
Figure 1: Schematic representation of the Xp11.22 deletions. The deletions of each subject are shown in relation to the position of Xp11.22 genes. The coordinates shown at the top of the figure are based on hg19. The RefSeq genes located in this critical region—CENPVL1, CENPVL2, GSPT2, and MAGED1—are shown in red. Genes depicted in blue are deleted in a subset of subjects 1–4, but not in our patient. Genes depicted in gray were not deleted in any subject.
Discussion
Recently, Grau and colleagues reported four males with Xp11.22 deletions associated with a distinct deletion syndrome.1 Our patient shares the same phenotype as these four males but with a 334 kb deletion that corresponds to the minimal region of overlap reported [Table 1]. This region encompasses two protein-coding genes: MAGED1 and GSPT2.
Both the MAGED1 and GSPT2 genes are ubiquitously expressed in the brain,4 and are conserved among vertebrates; however, the GSPT2 gene exhibits greater evolutionary conservation.5 Based on silico statistical methods such as the Exome Aggregation Consortium pLi score6 and the Residual Variation Intolerance Score,7 both genes exhibit intolerance to loss-of-function (LOF) mutations within the normal adult population. However, at least for the GSPT2 gene, there are no LOF single nucleotide variants (SNVs) in the gnomAD database6 or copy number deletions in the Database of Genomic Variants (DGV; http://dgv.tcag.ca/dgv/app/home).8 In contrast, four different copy number deletions encompassing the MAGED1 gene have been reported in the DGV database, and five healthy adult males with high impact LOF mutations have been reported in gnomAD [Table 2]. Of note, these SNVs affect all three known transcripts of this gene.
Table 2: Comparison between GSPT2 and MAGED1 by interrogating genomic databases.
|
Brain expression (GTEX) |
++ |
+++ |
|
Conservation (phyloP100wayAll) |
+++ |
++ |
|
RVIS (genic intolerance) |
-0.07 (48.12%) |
-0.54 (20.54%) |
|
ExAC pLi |
0.90 |
0.98 |
|
DGV deletions encompass the gene |
Absent |
Yes (nsv6906, esv2664331, esv3573968, esv3558875) |
|
GnomAD high impact mutations (males) |
Absent |
Five LOF variants |
RVIS: Residual Variation Intolerance Score; ExAC: Exome Aggregation Consortium; DGV: Database of Genomic Variants; LOF: loss-of-function; SNV: single nucleotide variant; VUS: variant of uncertain significance.
GSPT2 consist of one exon and encodes one known transcript. It encodes for peptide chain release factor 3b (eRF3B), one of the classic translation factor GTPase family. It is involved in the final step of protein synthesis where translation ends in response to the termination codons (UAA, UAG, and UGA). While GSPT1, which encodes for eRF3a, is expressed in every tissue. GSPT2 is highly expressed in the brain among other tissues in humans and mice.4,9 The effects of GSPT2 deficiency on central nervous system function have not been demonstrated in humans or studied in mice yet.
Conclusion
In summary, the phenotype of our patient is consistent with those previously reported cases of deletions involving MAGED1 and GSPT2. Interrogation of different public genomic databases indicates that GSPT2 is likely to be indispensable for brain function. Further understanding of the molecular function of this gene and its effect when knocked out in mouse and human cell lines is required.
Disclosure
The authors declared no conflicts of interest.
references
- 1. Grau C, Starkovich M, Azamian MS, Xia F, Cheung SW, Evans P, et al. Xp11.22 deletions encompassing CENPVL1, CENPVL2, MAGED1 and GSPT2 as a cause of syndromic X-linked intellectual disability. PLoS One 2017 Apr;12(4):e0175962.
- 2. Ropers H-H. X-linked mental retardation: many genes for a complex disorder. Curr Opin Genet Dev 2006 Jun;16(3):260-269.
- 3. Thiselton DL, McDowall J, Brandau O, Ramser J, d’Esposito F, Bhattacharya SS, et al. An integrated, functionally annotated gene map of the DXS8026-ELK1 interval on human Xp11.3-Xp11.23: potential hotspot for neurogenetic disorders. Genomics 2002 Apr;79(4):560-572.
- 4. Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al; GTEx Consortium. The genotype-tissue expression (GTEx) project. Nat Genet 2013 Jun;45(6):580-585.
- 5. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010 Jan;20(1):110-121.
- 6. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al; Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016 Aug;536(7616):285-291.
- 7. Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 2013;9(8):e1003709.
- 8. MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 2014 Jan;42(Database issue):D986-D992.
- 9. Chauvin C, Salhi S, Le Goff C, Viranaicken W, Diop D, Jean-Jean O. Involvement of human release factors eRF3a and eRF3b in translation termination and regulation of the termination complex formation. Mol Cell Biol 2005 Jul;25(14):5801-5811.