The clinical manifestations of Hashimoto’s encephalopathy (HE) include an altered level of consciousness, tremor, myoclonus, stroke-like episodes, and seizures. Seizures have been documented in 66% of patients with HE.1 Treatment consists of corticosteroids, which results in remarkable recovery thus giving this syndrome the term “steroid-responsive encephalopathy”.1
Case report
A 38-year-old woman was transferred to Sultan Qaboos University Hospital with a diagnosis of refractory status epilepticus (SE). At the time of presentation, she was eight weeks pregnant. She presented with a two-day history of fever and abdominal pain followed by a one-day history of depressed level of consciousness and generalized tonic-clonic seizures. There was no family history of seizure disorder, thyroid, or autoimmune disease. She progressed to generalized SE, which required intubation and mechanical ventilation.
She was started on intravenous (IV) anesthetics and phenytoin. Her temperature at initial examination was 38 oC. There were no signs of meningeal irritation, and her pupils were mid-position and reactive. An obstetrical examination revealed an empty sac consistent with an abortion.
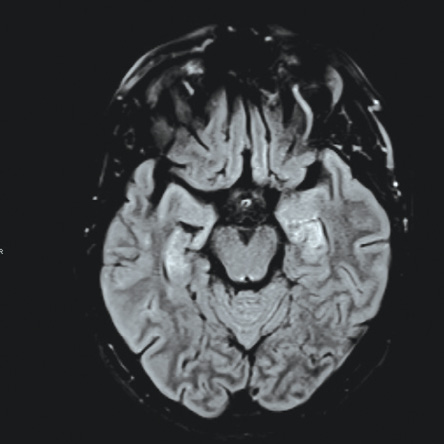
Figure 1: Magnetic resonance imaging showing pattern of bilateral symmetric increased signal hyperintensity of the hippocampal formation. There is increased signal intensity within the caudate nuclei and lentiform nuclei, which is associated with diffusion restriction.
Her investigations revealed normal complete blood count, serum electrolytes, and calcium profile. Cerebrospinal fluid showed three white blood cells, elevated proteins (0.96 g/L), normal glucose, and negative cultures. Computed tomography (CT) of the brain was normal while CT venography excluded venous sinus cerebral thrombosis. Magnetic resonance imaging (MRI) showed hyperintensities in the hippocampal region as well as bilateral basal ganglia [Figure 1]. An electroencephalogram (EEG) on admission showed continuous generalized frontally dominant rhythmic epileptiform discharges consistent with SE [Figure 2]. This pattern was resistant to sensory stimuli and would occasionally, and transiently, responds to IV midazolam. Thyroid peroxidase antibodies were positive (222 IU/mL). Another autoimmune workup was negative (including anti-Hu, anti-NMDA, anti-GAD65, ANA, and antiphospholipid). Viral screen was also negative including herpes simplex virus, polymerase chain reaction, and West Nile virus.
The patient was initiated on IV acyclovir empirically (until the exclusion of herpes encephalitis) with multiple sequential anticonvulsants including phenytoin, lamotrigine, levetiracetam, sodium valproate, clobazam, phenobarbital, and lacosamide. However, they failed to completely suppress the seizures. After 48 hours of admission, she continued to have breakthrough uncontrollable seizures. Multiple IV anesthetics were used, including midazolam, propofol, ketamine, and thiopentone. Burst suppression was initially achieved, but seizures recurred during IV anesthetics withdrawal.
On day two after admission, IV methylprednisolone 1 g was initiated for five days. There was no notable response. IV immunoglobulin was also given for five days. Five sessions of plasmapheresis were started. There was still failure of seizure control with episodes of two to three tonic-clonic seizures per hour. Eventually, the course was complicated with pseudomonal sepsis with multiple organ failure. She died on day 18 after admission.
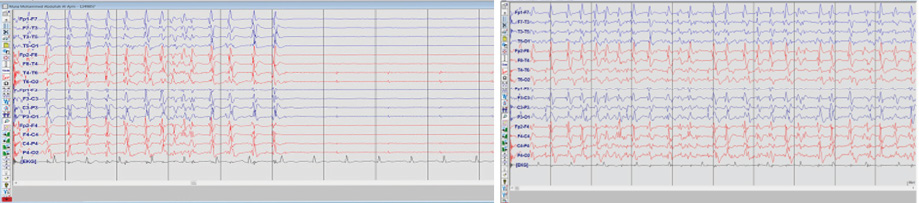
Figure 2: Electroencephalogram of status epilepticus showing continuous generalized, fairly rhythmic epileptiform discharges. The discharges were not responsive to any sensory stimulation.
Discussion
The above case highlights a fatal outcome of SE secondary to HE. We reviewed the literature for cases of SE secondary to HE with their findings and outcome [Table 1]. HE is considered a steroid-responsive disease with the overall outcome good even in the setting of SE.10,11 Chaigne et al,11 reported six cases of patients with SE admitted to intensive care, and all had an overall good outcome. However, there were two reported cases that showed severe refractoriness demonstrating that the disease course can have a resistant form that does not respond to immunotherapy.7,12
SE occurs in 12% of patients with HE.13 The diagnosis in these cases is challenging as SE has a variety of causes.14 Immunological causes are the most challenging to diagnose and treat.14 The most common immunological disorders are paraneoplastic syndromes, HE, and anti-NMDA receptor encephalitis.14
The hallmark of diagnosing HE is the presence of antithyroid antibodies.1,15 The most common antibody identified is antithyroperoxidase (anti-TPO) formerly known as antimicrosomal antibodies.15 It is found in most patients with HE. The second antibody commonly found in HE is antithyroglobulin (anti-TG),15 which is found to a lesser extent. Anti-alpha anolase is another antibody that can be diagnostic of HE.16 In one study, five out of six patients with HE were positive compared with 25 control patients with encephalopathy.16 In our patient, the diagnosis of HE was based on the exclusion of all other causes and the presence of anti-TPO with a level of 222 IU/mL. It is not clear in the literature what level is considered abnormal or diagnostic, as there is variability in the sensitivity and reference range.17 The reference range in our laboratory is 0–50 IU/mL. Other paraneoplastic and autoimmune antibodies were negative.
The cerebrospinal fluid findings in most cases with SE secondary to HE is normal as evident by Table 1. Few cases showed high protein level with no leukocytes.8,9 Most patients had normal MRI finding.3-6
EEG findings in our patient consisted of bifrontal spikes and waves, the most commonly identified pattern in reported cases.18 In a study that looked at EEG findings in HE in general, there were several findings including generalized abnormalities like slowing, triphasic waves, and periodic sharp waves.18 Focal temporal slowing was also demonstrated in some patients. Those EEG findings also varied within patients.18
The mainstay of treatment of HE is corticosteroids. The majority of cases show remarkable response with good outcome after treatment with pulse methylprednisolone.13,15 Other modalities of immunotherapy used in HE are plasmapheresis and IV immunoglobulin.5,19 However, although there are cases where those modalities were used, the long-term outcome is uncertain.19 In our case, there was progression of SE despite the use of IV immunoglobulin and plasmapheresis. This was most likely a state of SE refractoriness rather than a failure of a treatment targeting HE.
Table 1: Summary of reported cases of Hashimoto’s encephalitis presenting with status epilepticus, investigations, and treatments.
|
Tsai et al2 |
16-year-old girl presents with confusion |
Anti-TPO,
Anti-TG titers:
1:1 600 |
Normal |
Right hyperintense lesions right medial temporal |
High amplitude delta (Right parietal-temporal) |
Methylprednisolone |
Recovered |
|
Monti et al3 |
51-year-old with Hashimoto thyroiditis present with convulsive seizures |
Anti-TPO: 349.4 IU/mL
Anti-TG: > 500 IU/mL |
Normal |
Normal |
Bilateral frontal spikes and waves |
Methylprednisolone
1 g × 2 days, 500 mg × 3 days. |
Recovered |
|
Monti et al3 |
66-year-old admitted with non-convulsive status |
Anti-TPO: 643.9 IU/mL
Anti-TG: 126.9 IU/mL |
Normal |
Normal |
Bilateral frontal spikes and waves |
Methylprednisolone
1 g × 2 days, 500 mg × 3 days. |
Recovered |
|
McGinley et al4 |
42-year-old admitted with convulsive status |
Anti-TPO titre: 1:25 600
Anti-TG: negative |
Normal |
Normal |
Bilateral slow waves |
Dexamethasone |
Recovered |
|
Bektas et al5 |
12-year-old presents with behavioral changes and convulsive status |
Anti-TPO:
725 IU/mL
Anti-TG: 100 IU/mL |
Normal |
Unavailable |
Bilateral frontal spikes |
IVIG followed by
plasmapheresis and steroids |
Recovered |
|
McKeon et al6 |
61-year-old with absence status epilepticus |
Anti-TPO titer: 1:6 400
Anti-TG: unavailable |
Normal |
Normal |
Bilateral slow spikes and waves |
Steroids × 6 days |
Recovered |
|
Striano et al7 |
27-year-old woman presents with myoclonus then status epilepticus |
Anti-TPO: 1 781
IU/mL
Anti-TG:
127.5 IU/mL |
Raised protein 55 mg/dL,
two oligoclonal bands |
Unavailable |
Bifrontal theta
stimulus-induced myoclonus |
Methylprednisolone |
Died,
uncontrolled seizures |
|
Ferlazzo et al8 |
41-year-old male with convulsive status epilepticus |
Anti-TPO:
3 107 IU/mL
Anti-TG:
36 569 IU/mL |
Increased proteins |
Normal |
Generalized epileptic discharges |
IV methylprednisolone |
Recovered |
|
Canton et al9 |
17-year-old female with generalized tonic-clonic seizures |
Anti-TG:
638 IU/ml |
High protein 85 g/dL |
Normal |
Frontal sharp waves |
Methylprednisolone |
Recovered |
|
Canton et al9 |
CSF: cerebrospinal fluid; EEG: electroencephalogram; MRI: magnetic resonance imaging; Anti-TPO: antithyroperoxidase; Anti-GT: antithyroglobulin; IV: intravenous; IVIG: intravenous immunoglobulin.
Conclusion
HE should be considered in patients admitted with SE. Thyroid antibodies should be among the panel of investigation. While the overall prognosis is good, this case report demonstrates that severe progressive encephalopathy, particularly with refractory SE in such patients, may have a fatal outcome. However, prompt treatment with corticosteroids should be initiated before the period of super-refractoriness sets in.
Disclosure
The authors declared no conflicts of interest.
references
- 2. Ferracci F, Bertiato G, Moretto G. Hashimoto’s encephalopathy: epidemiologic data and pathogenetic considerations. J Neurol Sci 2004 Feb;217(2):165-168.
- 3. Tsai M-H, Lee L-H, Chen S-D, Lu C-H, Chen M-T, Chuang Y-C. Complex partial status epilepticus as a manifestation of Hashimoto’s encephalopathy. Seizure 2007 Dec;16(8):713-716.
- 4. Monti G, Pugnaghi M, Ariatti A, Mirandola L, Giovannini G, Scacchetti S, et al. Non-convulsive status epilepticus of frontal origin as the first manifestation of Hashimoto’s encephalopathy. Epileptic Disord 2011 Sep;13(3):253-258.
- 5. McGinley J, McCabe DJ, Fraser A, Casey E, Ryan T, Murphy R. Hashimoto’s encephalopathy; an unusual cause of status epilepticus. Ir Med J 2000 Jun;93(4):118-119.
- 6. Bektas Ö, Yılmaz A, Kendirli T, Sıklar Z, Deda G. Hashimoto encephalopathy causing drug-resistant status epilepticus treated with plasmapheresis. Pediatr Neurol 2012 Feb;46(2):132-135.
- 7. McKeon A, McNamara B, Sweeney B. Hashimoto’s encephalopathy presenting with psychosis and generalized absence status. J Neurol 2004 Aug;251(8):1025-1027.
- 8. Striano P, Pagliuca M, Andreone V, Zara F, Coppola A, Striano S. Unfavourable outcome of Hashimoto encephalopathy due to status epilepticus. One autopsy case. J Neurol 2006 Feb;253(2):248-249.
- 9. Ferlazzo E, Raffaele M, Mazzù I, Pisani F. Recurrent status epilepticus as the main feature of Hashimoto’s encephalopathy. Epilepsy Behav 2006 Feb;8(1):328-330.
- 10. Cantón A, de Fàbregas O, Tintoré M, Mesa J, Codina A, Simó R. Encephalopathy associated to autoimmune thyroid disease: a more appropriate term for an underestimated condition? J Neurol Sci 2000 May;176(1):65-69.
- 11. Kothbauer-Margreiter I, Sturzenegger M, Komor J, Baumgartner R, Hess CW. Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment. J Neurol 1996 Aug;243(8):585-593.
- Chaigne B, Mercier E, Garot D, Legras A, Dequin PF, Perrotin D. Hashimoto’s encephalopathy in the intensive care unit. Neurocrit Care 2013 Jun;18(3):386-390.
- 12. Duffey P, Yee S, Reid IN, Bridges LR. Hashimoto’s encephalopathy: postmortem findings after fatal status epilepticus. Neurology 2003 Oct;61(8):1124-1126.
- 13. Chong JY, Rowland LP, Utiger RD. Hashimoto encephalopathy: syndrome or myth? Arch Neurol 2003 Feb;60(2):164-171.
- 14. Tan RY, Neligan A, Shorvon SD. The uncommon causes of status epilepticus: a systematic review. Epilepsy Res 2010 Oct;91(2-3):111-122.
- 15. Mocellin R, Walterfang M, Velakoulis D. Hashimoto’s encephalopathy : epidemiology, pathogenesis and management. CNS Drugs 2007;21(10):799-811.
- 16. Fujii A, Yoneda M, Ito T, Yamamura O, Satomi S, Higa H, et al. Autoantibodies against the amino terminal of α-enolase are a useful diagnostic marker of Hashimoto’s encephalopathy. J Neuroimmunol 2005 May;162(1-2):130-136.
- 17. Sinclair D. Analytical aspects of thyroid antibodies estimation. Autoimmunity 2008 Feb;41(1):46-54.
- 18. Henchey R, Cibula J, Helveston W, Malone J, Gilmore RL. Electroencephalographic findings in Hashimoto’s encephalopathy. Neurology 1995 May;45(5):977-981.
- 19. Olmez I, Moses H, Sriram S, Kirshner H, Lagrange AH, Pawate S. Diagnostic and therapeutic aspects of Hashimoto’s encephalopathy. J Neurol Sci 2013 Aug;331(1-2):67-71.