Thrombocytosis in children is either attributed to intrinsic dysregulation of megakaryocytic proliferation and platelet production, known as primary thrombocytosis or develops in the context of other diseases (called secondary or reactive thrombocytosis). Secondary thrombocytosis is more common and is associated with iron deficiency anemia, infection, chronic inflammatory disorders (including inflammatory bowel disease), tissue injury, drugs, and certain neoplasms.1 The incidence of secondary thrombocytosis is highest in neonates and infants and generally, declines with age.2 On the other hand, primary thrombocytosis is rare and occurs in two settings: essential thrombocythemia (ET) and familial thrombocytosis (FT).1 A subset of patients with ET harbor a mutation in the Janus Kinase gene (JAK2), while in FT there is a mutation in thrombopoietin gene (TPO) or in its receptor (c-MPL) that leads to constitutive activation.3
Case Report
In 2007, a two-year-old female child was incidentally found to have isolated thrombocytosis on hemogram test. She was healthy, and there was no history of thrombocytosis in her parents or siblings. Secondary causes of thrombocytosis were excluded. High platelet count persisted on follow-up visits, while white blood cell count and hematocrit level were normal. Initial bone marrow examination showed unremarkable morphology and both JAK2V617F and BCR-ABL1 mutations were negative by polymerase chain reaction on peripheral blood.
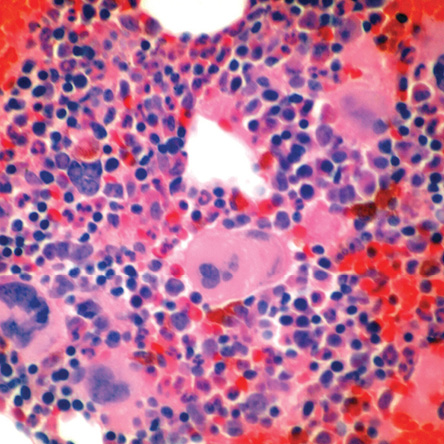
Figure 1: Bone marrow trephine biopsy revealed crowded megakaryocytes with hyperlobated nuclei (hematoxylin and eosin stain, magnification = 400 ×).
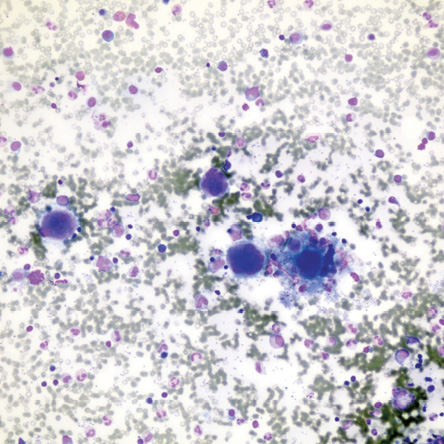
Figure 2: Bone marrow aspirate smear demonstrates increased number of megakaryocytes (Leishman stain, magnification = 200 ×).
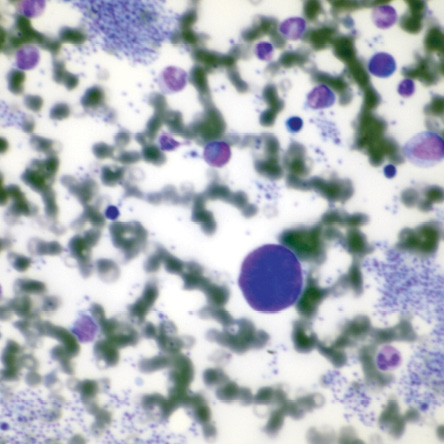
Figure 3: Bare megakaryocytic nuclei and platelet clumps are common features of essential thrombocythemia (Leishman stain, magnification = 400 ×).
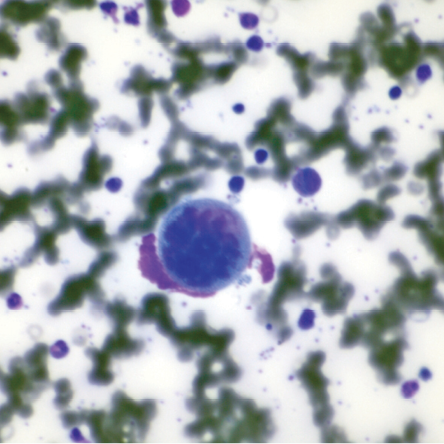
Figure 4: A megakaryocyte with “stag horn” nucleus is characteristic of essential thrombocythemia (Leishman stain, magnification = 400 ×).
The patient did not receive therapy until the age of four when she was initiated on a once-daily aspirin 100 mg. However, after a few months, she complained of headache and her platelets count increased progressively from 600 000 to 1 100 000/L within two months. Treatment was switched to hydroxyurea (35 mg/kg four times per week), and her platelets count normalized. She was followed-up on a monthly basis over a period of six years. Her clinical course was significant only for two episodes of minor skin bruises, epistaxis, and one event of mild elevation of liver enzymes that was attributed to hydroxyurea. Her dose was lowered, and the patient remained symptom-free.
In 2015, at the age of 10, her platelet count increased again. A second bone marrow specimen showed megakaryocytic proliferation [Figures 1 and 2], with typical features of ET, such as naked megakaryocytic nuclei [Figure 3] and nuclear hyperlobation [Figure 4]. Repeat JAK2 and BCR-ABL1 mutation studies were negative. Hydroxyurea dose was increased to 35 mg/kg daily, and patient compliance was assured.
Her platelet count decreased and remained between 600 000–700 000/L. No hepatosplenomegaly was detected during the entire course of the disease.
Discussion
ET is a chronic myeloproliferative neoplasm that primarily involves the megakaryocytic lineage, characterized by sustained thrombocytosis
(> 450 000/L) in peripheral blood, increased numbers of large mature megakaryocytes in the bone marrow, and clinical episodes of thrombosis and/or hemorrhage. The diagnosis requires the absence of significant increase or left-shift of neutrophil granulopoiesis or erythropoiesis, no meeting of World Health Organization criteria of polycythemia vera, primary myelofibrosis, BCR-ABL1+ chronic myelogenous leukemia, myelodysplastic syndrome, other myeloid neoplasms, and no evidence of reactive thrombocytosis.4 Hence, ET is a diagnosis of exclusion, and many clinical diseases must be ruled out before establishing the diagnosis. The incidence of ET is estimated to be 0.6–2.5 per 100 000 per year and most patients are between 50–60 years old.4 The incidence in children is rare, estimated to be 1 per 10 million,5 with around 100 cases reported in the literature.6
ET is associated with a point mutation in JAK2V617F (JAK2) gene in around half of adult cases, thrombopoietin receptor (c-MPL) in 3–5% or calreticulin (CALR) genes.4 However, this association is less frequent in children, suggesting an alternative mutational pathway, or even questions the clonal nature of most ET cases in children.5
Clinically, the median age of presentation of ET in children is eight years (range six months to 16 years). The majority of patients are asymptomatic at presentation. Headache and epistaxis are the most common symptoms. Children with ET are reported to have a lower risk of major vascular complications compared to adults, with a rate of 11.1–13.0%, which is correlated with the presence of clonal mutations and/or very high platelet count. A major bleeding event is also rare and was not described in two major series. Symptoms or even thrombotic events might develop without an excessively high platelet count. Platelets function is characteristically abnormal in ET, and the median count ranges between 1 100 000–1 700 000/L. Splenomegaly is present in around one-third of the patients.6,7 Typical bone marrow morphologic features include normal cellularity for the age and megakaryocytic proliferation, where megakaryocytes are abnormally large, mature with hyperlobated nuclei.4 However, as in the initial biopsy in our patient, the megakaryocytes in pediatric ET are often morphologically unremarkable, and it is unclear whether the status of JAK2 mutation affects morphology.6 Thus, the normal bone marrow morphology does not rule out ET in children, and a follow-up biopsy may confirm the diagnosis and evaluate disease progression. Both myeloid and erythroid lineages are unremarkable, and the stroma shows no or little reticulin fibrosis.4 The rate of progression to bone marrow fibrosis or transformation into acute leukemia is rare in children.8
Survival of ET at all age groups is not different from an age- and gender-matched normal population in the first 15 years after diagnosis. In adult patients, risk factors for major complications are age > 60 years and previous history of thrombosis. Other proposed risk factors include the presence of cardiovascular disease, leukocytosis, and positive JAK2 mutation, while extreme thrombocytosis (≥ 1 000 × 109/L) is associated with bleeding tendency. Clinical studies have shown that cytoreductive therapy reduced the rate of thrombosis and platelets count, and aspirin is useful in low-risk patients. However, the reliability of this guideline for pediatric patients is less clear, and children with ET should receive conservative management. Aspirin is given in the setting of microcirculatory symptoms or thrombosis, but is contraindicated when platelets count is > 1 500 000/L, if there was a previous bleeding episode, or at very young age due to the rare but calamitous possibility of Reye syndrome. Cytoreductive drugs should be used only as a last resort. Hydroxyurea (starting dose 15 mg/kg per day) is well-tolerated, effective in lowering platelets count and thrombotic events and carry little risk of leukemic transformation with long-term use. Other suitable agents include interferon alpha and anagrelide with variable response. Our patient was considered low-risk during the entire course of the disease. Adding aspirin did not prevent symptoms of microcirculatory disturbance, while hydroxyurea was beneficial in reducing platelets count and preventing major complications over six years. Adjusting the dose and assuring compliance were important points during management.9,10
Conclusion
ET is a rare cause of thrombocytosis in the pediatric population. The diagnosis and management can be challenging. Compared to adults, ET in children is characterized by normal or little morphologic abnormality in megakaryocytes, less frequency of clonal mutations, lower vascular complications, and longer disease duration. It may be reasonable to use specific risk stratification for children to guide management, though further studies are needed to confirm risk factors that are specific for children.
Disclosure
The authors declared no conflicts of interest.
references
- 1. Deaconu A, Bica C, Bulucea D. Reactive Thrombocytosis in Pediatric Pathology. Medicina (B Aires) 2014;21:51-54.
- 2. Indolfi G, Catania P, Bartolini E, Azzari C, Massai C, Poggi GM, et al. Incidence and clinical significance of reactive thrombocytosis in children aged 1 to 24 months, hospitalized for community-acquired infections. Platelets 2008 Sep;19(6):409-414.
- 3. Skoda R, Prchal JT. Lessons from familial myeloproliferative disorders. Semin Hematol 2005 Oct;42(4):266-273.
- 4. Thiele J, Kvasbicka HM, Orazi A, Tefferi A, Gisslinger H. Essential thrombocythaemia. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon: IARC; 2008. p. 48-50.
- 5. Fu R, Zhang L, Yang R. Paediatric essential thrombocythaemia: clinical and molecular features, diagnosis and treatment. Br J Haematol 2013 Nov;163(3):295-302.
- 6. Robert B. Lorsbach. Neoplastic Hematopathology: Myeloproliferative neoplasms. In: Maria A. Proytcheva, ed. Diagnostic Pediatric Hematopathology. 1st ed. New York: Cambridge University Press; 2011. p. 225-228.
- 7. Randi ML, Geranio G, Bertozzi I, Micalizzi C, Ramenghi U, Tucci F, et al. Are all cases of paediatric essential thrombocythaemia really myeloproliferative neoplasms? Analysis of a large cohort. Br J Haematol 2015 May;169(4):584-589.
- 8. Giona F, Teofili L, Moleti ML, Martini M, Palumbo G, Amendola A, et al. Thrombocythemia and polycythemia in patients younger than 20 years at diagnosis: clinical and biologic features, treatment, and long-term outcome. Blood 2012 Mar;119(10):2219-2227.
- 9. Harrison CN, Bareford D, Butt N, Campbell P, Conneally E, Drummond M, et al; British Committee for Standards in Haematology. Guideline for investigation and management of adults and children presenting with a thrombocytosis. Br J Haematol 2010 May;149(3):352-375.
- 10. Barbui T. How to manage children and young adults with myeloproliferative neoplasms. Leukemia 2012 Jul;26(7):1452-1457.