|
Abstract
A 33-year old Malay woman with undiagnosed systemic lupus erythematosus (SLE) and other comorbids presented with acute glomerularnephritis, hypertensive emergency and later acute confusional state. Cranial MRI revealed features consistent with posterior reversible encephalopathy syndrome (PRES).
Keywords: Posterior reversible encephalopathy syndrome (PRES); Systemic lupus erythematosus.
Introduction
Posterior reversible encephalopathy syndrome (PRES) is a clinicoradiological entity that can be missed by many clinicians. A high index of suspicion should alarm a clinician to think of PRES when patients present with seizures, altered mental status, headaches and visual disturbances. To the best of our knowledge; neurologic or psychiatric abnormalities can be a manifestation of active systemic lupus erythematous (SLE), hence the condition can be under-diagnosed. Even though PRES in SLE patients has been reported in other parts of the world; so far none has been reported in Malaysia. We present a case of PRES in an undiagnosed SLE patient.
Case Report
A 33-years old Malay woman, single, presented to the Ophthalmology Unit with a history of blurring of vision, associated with headache, nausea and vomiting for a week. She was otherwise healthy prior to this presentation. The patient’s blood pressure at presentation was 160/110 mmHg. Fundoscopic examination showed bilateral papilloedema with difficulty to assess visual field as her visual acuity was down to finger counting. Other systemic examination was unremarkable. The patient was treated as hypertensive emergency with intravenous nitrate followed by labetolol. Her blood pressure was eventually controlled to approximately systolic of 120-140 mmHg and diastolic of 60-90 mmHg.
Computed tomography of the brain was normal and there was no evidence of left ventricular hypertrophy on electrocardiography. Laboratory investigations revealed serum creatinine of 162 umol/L, serum albumin of 18 umol/L, proteinuria (3+/hpf), with red blood cells (2+/hpf). Her lipid profile showed raised triglycerides (3.4 mmol/L) and low density lipoproteins (LDL: 8.9 mmol/L). Her antinuclear antibody (ANA) was positive with homogenous pattern 1:320 dilution with positive anti-dsDNA (qualitative). Her urine output was good. Based on the clinical presentation and laboratory results; the patient was diagnosed with glomerulonephritis complicating the underlying systemic lupus erythematous (SLE).
Over the ensuing days of hospitalization, the patient developed a confusional state despite normal blood pressure and presumptive diagnosis of sagittal sinus thrombosis was made in the presence of nephrotic syndrome. Cranial T2-weighted/fluid-attenuated inversion recovery (FLAIR) MRI showed scattered areas of increased signal intensity seen in the white matter of the left posterior occipitoparietal regions, low on T1 weighted; with no enhancement post-contrast (Fig. 1). Her state of confusion resolved within 24 hours though her visual disturbance had minimally improved without treatment.
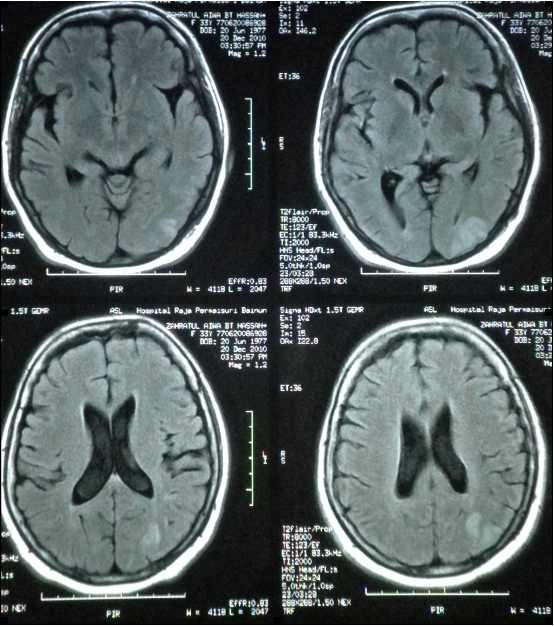
Figure 1: Cranial T2-weighted MRI showing hyperintense area in the left occipital area
Unfortunately, further treatment and assessment were not pursued as the patient insisted on seeking alternative medicine, therefore other auto antibodies pertaining to lupus including complement level were not sent. Patient stayed in the ward for less than a week and took self discharge before the anti-dsDNA results came into the clinical picture.
Discussion
Hincley et al. first described reversible posterior leukoencephalopathy in 1996, now most commonly known as posterior reversible encephalopathy syndrome.1 It can be a manifestation of a variety of disorders with neuroimaging changes suggestive of white matter edema, predominantly in the posterior regions of the brain. The common presentations are headache and altered conscious level, as well as visual problems including cortical blindness and seizures.1-3 This condition has been reported in hypertensive encephalopathy, pre-eclamptic toxemia, renal failure, immunosuppressant such as cyclosporine or high dose steroid, cytotoxic drugs and connective tissue diseases.1,4 The pathophysiology of PRES involves cerebral edema with diffusion of plasma proteins and cells into the extracellular space. However, the actual mechanism remains unclear. Two distinct theories have been proposed: the vasogenic and the cytotoxic theories.2,4
PRES has been well described in patients with established diagnosis of SLE with lupus nephritis. In SLE, patients are more prone to endothelial dysfunction, mainly due to its autoimmune or ischemic complications (vasculitis and thrombosis).4,5 In addition, the use of cytotoxic or immunosuppressants in these group of patients may worsen the vasogenic edema. Fluid retention and high blood pressure in lupus nephritis may also worsen the vasogenic edema. Both cytotoxic and vasogenic theories could explain the susceptibility of PRES amongst SLE patients.
The posterior aspect of the brain is commonly affected due to the differences in autonomic innervations between the anterior and posterior circulations.1 The posterior column has lesser sympathetic chain, and hence diminishes the ability to autoregulate well in the setting of sudden change in blood pressure, bodily fluid or biochemical components.
Typically, imaging shows T2-weighted and FLAIR bilateral subcortical and cortical hyperintensities of the white and grey matter with a predominantly posterior distribution.4,6 However, the involvement of other areas such as the frontal lobes, basal ganglia, thalamus and brainstem have also been reported. Diffusion-weighted imaging (DWI) and apparent diffusion coefficient (ADC) analysis reveal vasogenic cerebral edema in the majority of cases.4,6 Both DWI and ADC are used to differentiate vasogenic edema not caused by ischemic injury in acute setting. Low ADC value lesions are rarely present, and lesions with high ADC values are most often reversible.4,6 Those with decreased ADC values would progress to full blown infarction. Consequently, T2 weighted imaging, DWI signal changes and ADC values allow early differential diagnosis between PRES and acute cerebral infraction.2,4,6
The main differential diagnosis is bilateral ischemic stroke of the posterior cerebral artery territory. It would be crucial to distinguish PRES from a stroke as the latter does not involve rapid control of blood pressure. If hypertension complicates PRES, controlling the blood pressure with intravenous agent, to avoid permanent damage is warranted. In acute ischemic infarction however, blood pressure control is only mandatory when there is end organ damage such as hypertensive encephalopathy or acute pulmonary edema. PRES can also be mistaken for central venous sinus thrombosis, demyelinating disorders, lupus encephalitis, cerebral vasculitis and infectious or metabolic encephalopathy. Treatment must be instituted immediately. Parenteral antihypertensive treatment should be started with close blood pressure control, avoiding hypoperfusion as this could worsen the cerebral edema. Other supportive treatments such as hemodialysis are needed for fluid overloaded patients. If appropriate, the patients may need anticonvulsants and nursed in the intensive care unit.
The Oxford Journals (Nephrology, Dialysis and Transplant) have reported at least three cases of SLE patients who were diagnosed with PRES, noted to have Lupus Nephritis Class IV on renal biopsy. This case certainly illustrated the severity of the disease and despite rapid reduction of blood pressure; the patient’s vision did not fully recover. Hence, the term reversible can therefore be a misnomer as 50% of cases may have permanent deficit, especially with regards to vision.3,5 Her MRI could be repeated after 28 days to assess resolution of the edema, though no consensus on the actual length of time for a repeat MRI had been reported. In some cases, the MRI was repeated as early as 12 days and showed full recovery.4,5 In the current case, the patient certainly needed further treatment for her lupus nephritis in order to prevent progression to end stage renal disease. Her 24 hour urinary protein was 1096 mg/day and this value certainly warrants an urgent renal biopsy to be done. The standard treatment with methylprednisolone and cyclophosphamide would have been instituted.
As previously mentioned, PRES is usually under recognized amongst SLE patients. This is simply because the diagnosis of CNS lupus would have been made. In this case, the rapid rise of the patient’s blood pressure, together with renal involvement and the typical distribution of posterior-occipital region suggestive of edema on T2-weighted/FLAIR with normal MRA and MRV supported the diagnosis of PRES, though the radiologist did not make any comment on the DWI/ADC findings. Clinically, the patient improved after rapid stabilization of her blood pressure without any steroid being administered to the patient. In cerebral lupus however, the neuropsychiatric manifestation usually requires high dose immunosuppressive agents.
Conclusion
PRES is under recognized because of CNS abnormalities in SLE patients. It has great implications in patients who present with high blood pressure (95% of cases), renal involvement (91% of cases) or on immunosuppressive therapy (54% of cases).4 It requires both nephrologists and rheumatologists to intricately manage this group of delicate patients. Prompt diagnosis and treatment are therefore mandatory for full recovery of abnormalities.
Acknowledgements
The authors reported no conflict of interest and no funding was received for this work.
References
1. Hinchey J, Chaves C, Appignani B, Breen J, Pao L, Wang A, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med 1996 Feb;334(8):494-500.
2. Hagemann G, Ugur T, Witte OW, Fitzek C. Recurrent posterior reversible encephalopathy syndrome (PRES). J Hum Hypertens 2004 Apr;18(4):287-289.
3. Morelle J, Cosnard G, Jadoul M, Kanaan N. Transient blindness & seizures in severe lupus nephritis. Nephrol Dialysis Transplant Plus 2009;2:331-332.
4. El Karoui K, Le Quintrec M, Dekeyser E, Servais A, Hummel A, Fadel F, et al. Posterior reversible encephalopathy syndrome in systemic lupus erythematosus. Nephrol Dial Transplant 2008 Feb;23(2):757-763.
5. The Medline reference is Kur JK, Esdaile JM. Posterior reversible encephalopathy syndrome–an underrecognized manifestation of systemic lupus erythematosus. J Rheumatol 2006 Nov;33(11):2178-2183.
6. Schwartz RB, Mulkern RV, Gudbjartsson H, Jolesz F. Diffusion-weighted MR imaging in hypertensive encephalopathy: clues to pathogenesis. AJNR Am J Neuroradiol 1998 May;19(5):859-862.
|