|
Introduction
Systemic sclerosis is a multisystem disease characterized by inflammation and degeneration of integument, heart, lung, kidney, gastrointestinal tract, and synovium. Its etiology is not fully understood, though many occupational exposures are implicated, such as vinyl chloride, epoxy benzene, organic solvent and less commonly, silica exposure. In 1914, Bramwel first reported an association between ‘scleroderma’ and occupations associated with silica exposure.1 In 1957, Erasmus described an apparently high prevalence of progressive systemic sclerosis (PSS) in Witwatersrand gold miners exposed to dust containing a high percentage (±30%) of free silica compared with a large male non-mining hospital population which he used as a reference population.2 The only other epidemiological study based on the association between silicosis and PSS is one by Rodnan et al. who noted that of the 60 male patients diagnosed to be suffering from PSS in the period of 1955-1956, 26 had been exposed to "prolonged and heavy exposure to silica dust."3
Devulder et al. discussed the association and presented a bibliography up to 1977.4 They suggested that the eponym "Erasmus syndrome" be used when referring to the co-existence of silicosis and PSS. The first case of silicosis-induced systemic sclerosis in India was published by N Khanna et al. in 1997.5 This report describes a case of a 26-year-old Indian male who developed diffuse parenchymal lung disease due to silicosis and systemic sclerosis after 6 years of exposure to silica dust.
Case Report
A 26-year-old nondiabetic, normotensive male presented at the chest OPD on 9/01/2012 with progressive dyspnea for the last 4 years, along with persistent dry cough for the same duration. There was one episode of streaky hemoptysis, but no history of any wheezing. The patient also complained of tightening of the skin on his hands, face, and perioral region for the last 2 years. There was also associated arthralgia involving both upper and lower limbs, low-grade intermittent fever and bluish discoloration of hands on exposure to cold, suggestive of Raynaud’s phenomenon lasting for approximately the same duration. There was no history of any dysphagia and there was no significant medical or family history. He worked as a stone crusher in Delhi for 1 year, and stone masonry for 5 years (2002-2008). After that, he quit his job due to severity of dyspnea.
On examination, there was loss of wrinkling over the forehead, microstomia, thickening and pigmentation of skin on the hands and forearm, sclerodactyly, and finger clubbing with loss of finger pad substance. Right supraclavicular lymph node was palpable, 3 cm in diameter, nontender, firm with normal overlying skin. Examination of respiratory system revealed bilateral end-inspiratory fine crepitations with rhonchi. High- resolution CT of the thorax revealed diffuse nodular lesions mostly involving the upper and middle lobes with conglomeration of nodules in a few areas (Fig. 1). There was extensive egg-shell calcification of mediastinal and hilar lymph nodes (Fig. 2). Pulmonary function test revealed severe restriction with FVC of 1.04 litres (25% predicted). Six-minute walk test showed a 6MWD of 310 meters, but the patient desaturated from 97% to 86%.
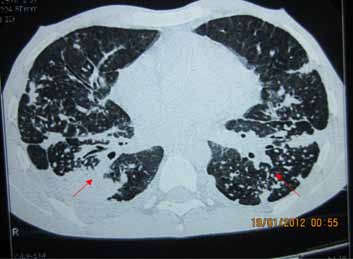
Figure 1: HRCT thorax showing diffuse nodular lesions mostly in the upper and middle lobe.
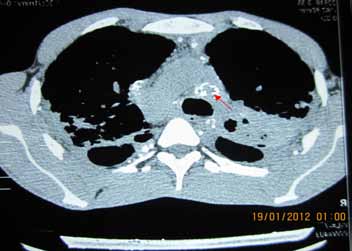
Figure 2: Egg-shell calcification seen in the mediastinal and hilar lymph nodes.
Anti Scl-70 antibody was strongly positive; ANA was also positive but RA factor was negative. Barium swallow was within normal limits. USG revealed early renal parenchymal disease though urinalysis, but liver and renal profiles were normal. Biopsy of the supraclavicular lymph node revealed reactive hyperplasia. Skin biopsy taken from the face showed epithelial thinning, increased melanin in the basal cell layer, dermal edema and fragmentation of collagen; overall, there was dermal shrinkage with reduced appendages and patchy lymphocytic infiltrate suggestive of scleroderma. (Fig. 3)
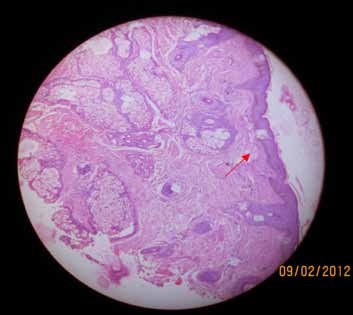
Figure 3: Skin biopsy from the face showing dermal shrinkage.
Discussion
Progressive systemic sclerosis is an autoimmune disease of unknown etiology with an estimated annual incidence of 19 new cases per million adults per year.6 It is characterized by three major processes: disease specific autoantibodies, organ fibrosis, and small vessel vasculopathy. There is a female preponderance towards the 30-50 years age group.7 Patients can be classified into two principal subsets defined largely by the pattern of skin involvement, as well as clinical and laboratory manifestations. Diffuse cutaneous SSc is associated with progressive skin induration, starting in the fingers and ascending from the distal to proximal extremities, the face, and the trunk. These patients are at risk of early pulmonary fibrosis and acute renal involvement. Patients with limited cutaneous SSc (lcSSc) generally have long-standing Raynaud's phenomenon before other manifestations of SSc appear. Skin involvement in lcSSc is slowly progressive and remains limited to the fingers (sclerodactyly), distal extremities, and face, but the trunk is not affected.8 The pathogenesis remains unknown but dermal fibroblast from involved skin accumulate type I, III and IV collagen, fibronectin and glycosaminoglycans at an increased rate.9
Silicosis is a fibrosing disease of the lung caused by inhalation, retention and pulmonary reaction to crystalline silica, exposure to which occurs as an occupational hazard in quarrying, mining, masonry and sand blasting. Pulmonary macrophages exposed to silica produce factors that cause chronic inflammation and fibroblastic proliferation leading to progressive fibrosis of the lungs.10 Exposure to silica has been reported as a cause of progressive systemic sclerosis, and it is postulated that chronic exposure to silica causes alterations in immunity, including alterations in soluble interleukin-2 (IL-2) receptors in affected patients.11 Increased lymphokines either stimulate collagen production or other cells like monocyte or mast cells to release factors that in turn induce collagen biosynthesis in fibroblasts. This is supported by the fact that increased levels of soluble IL-2 receptors are found in systemic sclerosis.9 The mean exposure time was 14.5 years (4-33 years) and intervals between exposure and development of silica was 24.4 years (4-45 years) in one study.12 Silicosis has also been seen in cases of short intense exposure (5-10 years), i.e., patients with accelerated silicosis are more prone to development of autoimmune disease. Our patient was exposed to silica dust for 6 years. Recent studies have also documented cases of females who underwent silicone implant for augmentation mammoplasty presenting with features suggestive of systemic sclerosis. There are also other studies which associate silica exposure with the incidence of bronchial anthracosis.13
Conclusion
Silica induced scleroderma is indistinguishable from idiopathic systemic sclerosis by epidemiological, clinical or immunological parameters, though the former expresses a higher prevalence of pulmonary involvement and anti Scl-70 antibody. However, the prognoses in both cases are similar. In our patient, radiological features were consistent with diffuse parenchymal lung disease due to silicosis, whereas clinical, serological and skin biopsy indicated the presence of scleroderma. Therefore, careful screening should be done in patients with silicosis along with systemic manifestation to rule out any associated connective tissue disorder.
Acknowledgements
The authors reported no conflict of interest and no funding was received for this work.
References
1. Bramwell B. Diffuse scleroderma: its frequency; its occurrence in stonemasons. Edinburgh Med J 1914;12:387-401.
2. Erasmus LD. Scleroderma in Gold-Miners on the Witwatersrand with Particular Reference to Pulmonary Manifestations. S Afr J Lab Clin Med 1957;3(3):209-231.
3. Rodnan GP, Benedek TG, Medsger TA Jr, Cammarata RJ. The association of progressive systemic sclerosis (scleroderma) with coal miners’ pneumoconiosis and other forms of silicosis. Ann Intern Med 1967 Feb;66(2):323-334.
4. Devulder B, Plouvier B, Martin JC, Lenoir L. The association: scleroderma-silicosis or Erasmus’ syndrome (author’s transl). Nouv Presse Med 1977 Oct;6(32):2877-2879.
5. Khanna N, D’Souza P, Sud A, Pandhi RK. Systemic sclerosis in a stone cutter. Indian J Dermatol Venereol Leprol 1997 Mar-Apr;63(2):111-113.
6. Mayes MD, Lacey JV Jr, Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum 2003 Aug;48(8):2246-2255.
7. Mayes MD, Revile JD. Epidemiology, Demographics and Genetics. In: Clements PJ, Furst DE, Editors. Systemic Sclerosis. 2nd Edition. Philadelphia: Lippincott Williams and Wilkins; 2004.p.1-15.
8. John Varga. Systemic sclerosis (scleroderma) and related disorders. In Anthony’s fauci, Eugene Braunwald et al, editors. Harrison’s principles of internal medicine.17th edition. New York: McGraw hills; 2008.p 2096-2106.
9. Kahaleh MB, LeRoy EC. Interleukin-2 in scleroderma: correlation of serum level with extent of skin involvement and disease duration. Ann Intern Med 1989 Mar;110(6):446-450.
10. Petsonk EL, Parker JE. Coal Workers’ Lung Disease and Silicosis. In Alfred P Fishman Editors, Fishman’s Pulmonary Disease and Disorders. 4th Editions. New York: 2008; 974-979.
11. Hayashi H, Maeda M, Murakami S, Kumagai N, Chen Y, Hatayama T, et al. Soluble interleukin-2 receptor as an indicator of immunological disturbance found in silicosis patients. Int J Immunopathol Pharmacol 2009 Jan-Mar;22(1):53-62.
12. Rustin MH, Bull HA, Ziegler V, Mehlhorn J, Haustein UF, Maddison PJ, et al. Silica-associated systemic sclerosis is clinically, serologically and immunologically indistinguishable from idiopathic systemic sclerosis. Br J Dermatol 1990 Dec;123(6):725-734.
13. Ghanei M, Aslani J, Peyman M, Asl MA, Pirnazar O. Bronchial anthracosis: a potent clue for diagnosis of pulmonary tuberculosis. Oman Med J 2011 Jan;26(1):19-22.
|
|