| |
Introduction
Very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency (MIM 201475) was first described in 1993.1,2 It is an autosomal recessive disorder due to deficiency in the VLCAD enzyme located on the inner mitochondrial membrane. The enzyme catalyses the initial step of mitochondrial ß-oxidation of long-chain fatty acids which is crucial for energy production in heart and skeletal muscles. The disease manifests a wide range of severity but often presents in early neonatal period with symptomatic hypoglycemia. Cardiac manifestations with or without hepatomegaly and hepatocellular dysfunction may subsequently develop at or around 2 to 5 months of age that may lead to death.3 In the early 1990s, most patients with VLCAD deficiency were reported to die rapidly following the onset of symptoms.4 The first report of successful treatment using medium chain triglycerides (MCT) based dietary formula and carnitine supplement was published in 1996 from USA.5 Subsequently, more cases were reported in the literature.6,7 In this report, we describe the first diagnosed and treated case of VLCAD deficiency with cardiomyopathy in Oman.
Case Report
YS was born full term by spontaneous vaginal delivery with normal birth weight. The parents are first degree cousins who had lost 2 previous children, the first one at 3 days of age with sudden unexplained death at home and the second at the age of 2 months with confirmed hypoglycemia and cardiomyopathy that was proven to be VLCAD deficiency based on tandem mass spectrometry (TMS) results.
In the immediate neonatal period, YS was observed to have one episode of borderline asymptomatic hypoglycemia (blood glucose: 2.7 mmol/L) that responded well to oral feeding. Based on family history, he was investigated with TMS which suggested the diagnosis of VLCAD deficiency by the presence of a very high peak of C14:1 carnitine 10.1 umol/L (normal range [NR]: 0 - 0.5) and high C14/C16 carnitine ratio of 0.41 (NR: 0 - 0.2). The family was advised to avoid fasting, observe symptoms of hypoglycemia during sickness, and dietary treatment based on MCT formula was ordered from abroad. Enzyme assay on skin fibroblast showed absent activity of VLCAD enzyme (0 nmol/min/mg; controls mean ± SD: 3.4 ± 0.9; range: 1.9 - 5.0) and hence confirmed the diagnosis.
Cardiac evaluation with echocardiography showed dilated left ventricle with mild mitral valve regurgitation. There was concentric left ventricular hypertrophy with intraventricular septal thickness of 7 mm (NR: 2.4 - 5.2 mm), z score of 4.57, and left ventricular posterior wall thickness of 7 mm (NR: 2.6 - 4.6 mm), and z score of 6.8. The ventricular ejection fraction was markedly reduced to 38%, with minimal pericardial effusion. Furosemide and captopril therapy were started. Nevertheless, the baby was readmitted to the hospital at 7 weeks of age with increasing floppiness, poor feeding and evidence of cardiac failure evidenced by tachypnea, tachycardia with gallop rhythm, hepatomegaly, and radiological findings of cardiomegaly. Other laboratory parameters revealed normoglycemia, normal serum creatine kinase, with normal liver function, arterial blood gas analysis and renal profile. Repeat echocardiography showed worsening of cardiac function with an increase in the pericardial fluid; hence, anti-failure medications were increased to maximum. The child was started on the MCT-based dietary formula treatment (Monogen® Nutricia, North America) containing 90% MCT and 10% long chain triglycerides (LCT), with subsequent improvement in the general condition and cardiac function. Reassessment at 6 months of age showed marked reduction in left ventricular dilation, disappearance of pericardial effusion, an increase in ejection fraction to 73%, with persistent left ventricular hypertrophy and trivial mitral valve regurgitation. Anti-failure medications were gradually weaned and the child has been off medications since two years of age.
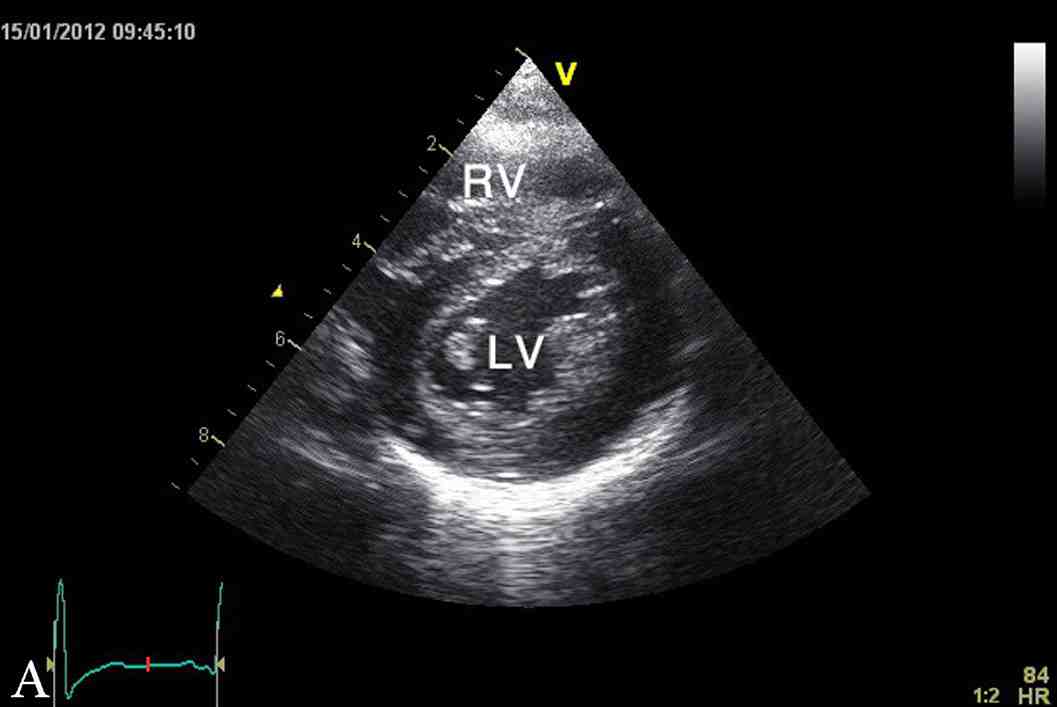
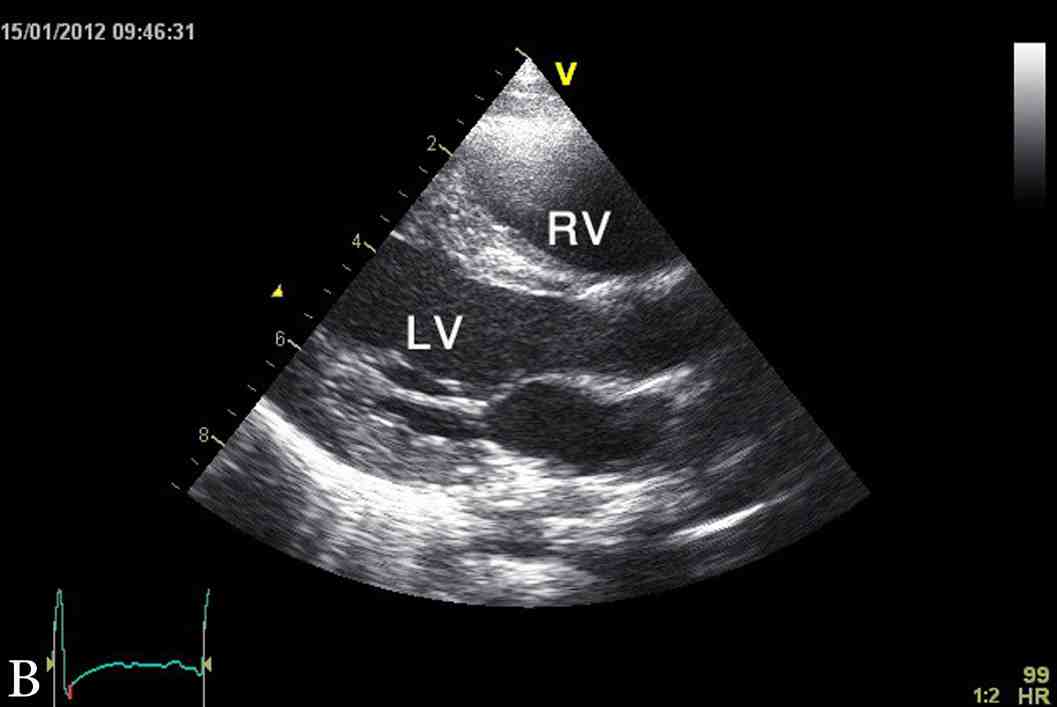
Figure 1: Latest echocardiographic parasternal long axis view (a) and short axis view (b) of the heart showing mild left ventricular hypertrophy. RV; right ventricle, LV; left ventricle.
The last echocardiography at 3.5 years of age showed mild concentric left ventricular hypertrophy with inter-ventricular septum of 9 mm (NR: 3.3 - 6.3 mm), z score of 5.6 and left ventricular posterior wall of 7 mm (NR: 3.4 - 6.4 mm), and z score of 2.8 (Fig. 1). At the time of this report, YS was a normal healthy four-year-old boy with appropriate weight and height, as well as normal cardio-respiratory functions and exercise tolerance. His height and weight are both at 90th percentile and had normal neurodevelopment. His current diet included Monogen milk formula, avoidance of all fatty diet, with low fat skimmed milk, MCT oil for general cooking, in addition to walnut oil (containing linoleic acid and α-linolenic acid) to substitute for the essential fatty acid requirement.
Discussion
Previously in Oman, undiagnosed cases of fatty acid oxidation defects might have died in early neonatal life with symptomatic hypoglycemia or sudden unexplained death. The family of our reported case had lost two previous children due to VLCAD deficiency; this time, as presented in our case, the patient’s condition was fortunate to have been recognized early by high-risk TMS screening. He showed rapid response to high concentration of MCT-based feeding formula. It was such a great joy to the family to see their child having normal growth and development in contrast to the previous two siblings.
VLCAD deficiency has been described to have three phenotypes: the severe early-onset form, hepatic/hypoketotic hypoglycemic form, and the adult onset myopathic form.8,9 In our patient, the phenotype fits with early onset presentation with biphasic course. Human VLCAD gene (ACADVL gene) is located at 17p13 region, where a number of mutations have been identified but genotype-phenotype correlation is not well established.10,11 Pre-symptomatic diagnosis of VLCAD deficiency in the newborn period may be suggested by acylcarnitine profile on TMS with high levels of long-chain acylcarnitines C16, C14, C14:1, and C12, with a C14:1 peak.12,13 While this may not be apparent in a healthy child who has not fasted, it is likely that patients with clinically significant VLCAD deficiency do exhibit abnormal biochemical markers in the immediate neonatal period. The diagnosis of fatty acid oxidation disorder is further strengthened by the presence of hypoketotic dicarboxylic aciduria. Skin biopsy fibroblast culture for direct enzyme assay is essential to forecast the clinical course.3 In the recent years, genetic diagnosis by DNA sequencing has been reported,11 however, it is still pending in our case. Identifying the mutation would be useful for genetic diagnosis in subsequent cases and planning for future pregnancies.
In VLCAD deficiency, there is a lack of cellular energy production as the primary mechanism of disease. Intravenous glucose is usually effective for the treatment of acute metabolic decompensation. For long-term management, a diet consisting of frequent meals that are high in carbohydrates, low in LCT, and supplemented with MCT oil has been recommended. Medium-chain fatty acids diffuse into mitochondria in a carnitine-independent manner and undergo ß-oxidation by enzymes with shorter chain length specificity.14 Although MCT-supplemented diet has been reported to prevent the development of cardiomyopathy and skeletal myopathy in fatty acid oxidation disorders, a recent study in VLCAD knock-out mice showed that long term MCT supplementation resulted in steatohepatitis and a phenotype like the metabolic syndrome,15 and this could also be true in humans. On the other hand, minimum intakes of LCT or daily supplements of essential fatty acids are mandatory.
Cardiomyopathy in infancy can be primary or secondary, with many children dying before the cause is identified.16 Secondary causes of hypertrophic cardiomyopathy include mitochondrial diseases, fatty acid oxidation defects, Noonan's syndrome, Friedreich's ataxia, and Fabry's disease. It is mandatory to explore the possible etiology since the natural history and prognosis are different, and initiating treatment may alter the disease course. Involvement of the heart in patients with VLCAD deficiency is well described in the literature.5-7 Affected patients usually develop features of hypertrophic cardiomyopathy with severe deterioration of cardiac function. These reports described improvement of cardiac status after commencing dietary therapy. In our patient, the MCT based formula resulted in dramatic improvement in general condition and cardiac function.
Conclusion
Early onset phenotype VLCAD deficiency is a severe and life-threatening defect of long-chain fatty acid oxidation that is often characterized by neonatal hypoglycemia followed by hypertrophic cardiomyopathy and cardiac failure. This can be reversed with early diagnosis and management by MCT-based formula. It is important that pediatricians, cardiologists, neonatologists, and all healthcare professionals are aware of this metabolic disorder. In most cases, the disease can easily be identified by TMS based acylcarnitine profile performed on a dry blood spot sample. This is one case along with many other metabolic disorders in the country that highlight the importance of initiating TMS based newborn screening program.
Acknowledgements
The authors reported no conflict of interest and no funding was received for this work.
References
1. Bertrand C, Largillière C, Zabot MT, Mathieu M, Vianey-Saban C. Very long chain acyl-CoA dehydrogenase deficiency: identification of a new inborn error of mitochondrial fatty acid oxidation in fibroblasts. Biochim Biophys Acta 1993 Jan;1180(3):327-329.
2. Aoyama T, Uchida Y, Kelley RI, Marble M, Hofman K, Tonsgard JH, et al. A novel disease with deficiency of mitochondrial very-long-chain acyl-CoA dehydrogenase. Biochem Biophys Res Commun 1993 Mar;191(3):1369-1372.
3. Vianey-Saban C, Divry P, Brivet M, Nada M, Zabot MT, Mathieu M, et al. Mitochondrial very-long-chain acyl-coenzyme A dehydrogenase deficiency: clinical characteristics and diagnostic considerations in 30 patients. Clin Chim Acta 1998 Jan;269(1):43-62.
4. Aoyama T, Souri M, Ushikubo S, Kamijo T, Yamaguchi S, Kelley RI, et al. Purification of human very-long-chain acyl-coenzyme A dehydrogenase and characterization of its deficiency in seven patients. J Clin Invest 1995 Jun;95(6):2465-2473.
5. Brown-Harrison MC, Nada MA, Sprecher H, Vianey-Saban C, Farquhar JR Jr, Gilladoga AC, et al. Very long chain acyl-CoA dehydrogenase deficiency: successful treatment of acute cardiomyopathy. Biochem Mol Med 1996 Jun;58(1):59-65.
6. Cox GF, Souri M, Aoyama T, Rockenmacher S, Varvogli L, Rohr F, et al. Reversal of severe hypertrophic cardiomyopathy and excellent neuropsychologic outcome in very-long-chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr 1998 Aug;133(2):247-253.
7. Pervaiz MA, Kendal F, Hegde M, Singh RH. MCT oil-based diet reverses hypertrophic cardiomyopathy in a patient with very long chain acyl-coA dehydrogenase deficiency. Indian J Hum Genet 2011 Jan;17(1):29-32.
8. Andresen BS, Olpin S, Poorthuis BJ, Scholte HR, Vianey-Saban C, Wanders R, et al. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet 1999 Feb;64(2):479-494.
9. Hoffman JD, Steiner RD, Paradise L, Harding CO, Ding L, Strauss AW, et al. Rhabdomyolysis in the military: recognizing late-onset very long-chain acyl Co-A dehydrogenase deficiency. Mil Med 2006 Jul;171(7):657-658.
10. Andresen BS, Bross P, Vianey-Saban C, Divry P, Zabot MT, Roe CR, et al. Cloning and characterization of human very-long-chain acyl-CoA dehydrogenase cDNA, chromosomal assignment of the gene and identification in four patients of nine different mutations within the VLCAD gene. Hum Mol Genet 1996 Apr;5(4):461-472.
11. Goetzman ES, Wang Y, He M, Mohsen AW, Ninness BK, Vockley J. Expression and characterization of mutations in human very long-chain acyl-CoA dehydrogenase using a prokaryotic system. Mol Genet Metab 2007 Jun;91(2):138-147.
12. Liebig M, Schymik I, Mueller M, Wendel U, Mayatepek E, Ruiter J, et al. Neonatal screening for very long-chain acyl-coA dehydrogenase deficiency: enzymatic and molecular evaluation of neonates with elevated C14:1-carnitine levels. Pediatrics 2006 Sep;118(3):1065-1069.
13. Al Riyami S, Al Maney M, Joshi SN, Bayoumi R. Detection of Inborn Errors of Metabolism using Tandem Mass Spectrometry among High-risk Omani Patients. Oman Med J 2012 Nov;27(6):482-485.
14. Hale DE, Bennett MJ. Fatty acid oxidation disorders: a new class of metabolic diseases. J Pediatr 1992 Jul;121(1):1-11.
15. Tucci S, Flögel U, Sturm M, Borsch E, Spiekerkoetter U. Disrupted fat distribution and composition due to medium-chain triglycerides in mice with a β-oxidation defect. Am J Clin Nutr 2011 Aug;94(2):439-449.
16. Jaiyesimi O, Kasem M. Clinical profiles and outcomes for Omani children with dilated cardiomyopathy seen in a regional referral hospital. Cardiol Young 2009 Apr;19(2):145-151.
|
|