Defects in human genes encoding peroxisomal proteins can result in different peroxisomal disorders with variable severity ranging from early lethality to subtle neurosensory abnormalities.1 Acyl- coenzyme A-binding domain-containing protein 5 (ACBD5) is a peroxisomal membrane protein with a C-terminal membrane-spanning region and an N-terminal cytosolic acyl-CoA binding domain. It is postulated to function as a peroxisomal membrane-bound receptor for very long-chain fatty acids (VLCFA) facilitating their transport into the peroxisome and subsequent β-oxidation.2,3 The absence of ACBD5 in human skin fibroblasts did not affect the biogenesis of peroxisomes, but impaired peroxisomal beta-oxidation of VLCFA, leading to elevation of the level of cellular phospholipids containing VLCFA without affecting peroxisomal biogenesis.3,4
The first report describing a biallelic pathogenic variant in the ACBD5 gene associated with retinal dystrophy and leukodystrophy was in 2013, involving three siblings from a consanguineous Saudi Arabian family.5 In 2017, Ferdinandusse et al,3 described a nine-year-old girl from the UAE who had retinal dystrophy, leukodystrophy, in addition to cleft palate and facial dysmorphism. In 2021, a 36-year-old Brazilian woman was reported with retinal dystrophy, leukodystrophy, and psychomotor regression.6 The seventh and most recent available report of this condition featured two Turkish sisters, aged five and nine years, with early-onset nystagmus, progressive motor decline and ataxia.7
This case series presents five Omani patients from four families to further emphasize the consistent clinical, radiological, and biochemical findings and expand the phenotypic spectrum of this relatively newly recognized disorder.
Case Reports
Case one (male, 10 years old)
This Omani boy is the eldest child of a non-consanguineous Omani couple. He was noted to have nystagmus at three months of age and spasticity at 14 months. Ophthalmic examination revealed horizontal, pendular nystagmus, sensitivity to light, severe visual impairment in both eyes, and mild compound myopic astigmatism. Fundus examination showed pale discs with attenuated vessels and pigmentary changes at the macula, which progressed to atrophy with mild tessellation of the background retina. Electroretinogram (ERG) showed severe cone-rod dysfunction. VLCFA profile showed elevated C26:0 of 1.38 umol/L (0.35–1.22), elevated C24/C22 ratio of 1.15 (0.74–1.02) and borderline C26/C22 of 0.02 (0.006–0.02), along with elevated pristanic and phytanic acids suggestive of a peroxisomal disorder. Other investigations included normal echocardiography and nerve conduction study. Intelligence Quotient test using the Stanford Binet 5th edition at the age of six years showed a total Intelligence Quotient of 65 corresponding to mild intellectual disability. Brain magnetic resonance imaging (MRI) showed demyelinated periventricular and deep white matter [Table 1, Figure 1: a–f]. Whole-exome sequencing (WES) of his younger affected sibling (Case two) identified a homozygous deletion in exons 7 and 8 in the ACBD5 gene. Upon assessment at the age of ten years, the patient had worsening mobility, intention tremor, and scanning speech. He was wheelchair-bound and was able to type only with great difficulty. He had full bladder and bowel control. He was microcephalic (head circumference at -3.6SD) with weight and height below the 2nd percentile (both at -2.7SD). Neurological examination was remarkable for central hypotonia, severe lower limb spasticity and scissoring, ankle joint contractures, and brisk deep tendon reflexes.
Table 1: Summary of the clinical features of five Omani patients with ACBD5-related retinal dystrophy with leukodystrophy.
|
Anthropometry
|
|
|
|
|
|
|
Head circumference
Weight
Height
|
-3.6SD
-2.7SD
-2.7SD
|
10th percentile
8th percentile
2nd percentile
|
-3SD
-4.8SD
-4.5SD
|
All at the 5th–10th percentile
|
2nd percentile
57 kg
171 cm
|
|
Genetic variant in ACBD5 gene
|
Homozygous deletion of exons 7–8
|
Homozygous deletion of exons 7–8
|
Homozygous deletion
of exons 7–8
|
Homozygous deletion of exon 4
|
Homozygous deletion of exon 4
|
|
Ophthalmological features
|
|
Age at presentation
|
2 years
|
2.5 years
|
18 months
|
16 months
|
17 years
|
|
Age of onset
|
3 months
|
3 months
|
5–6 months
|
3 months
|
< 6 months
|
|
Age of last ophthalmic assessment
|
9 years
|
4 years
|
7 years
|
7 years
|
21 years
|
|
Photophobia
|
+
|
+
|
+
|
+
|
+
|
|
Nystagmus (horizontal, pendular)
|
+
|
+
|
+
|
+
|
+
|
|
Visual acuity at presentation
(qualitative assessment)
|
OU <6/60*
|
OD: FF; CUSM
OS: FF; CUSM
|
OD: FF; CSM
OS: FF; CSM
|
OU: <6/60*
|
OU: <6/60*
|
|
Vision when last seen
|
OU <6/60
|
OU FF; CUSM
|
OU< 6/60*
|
OU: < 6/60
|
OU: <6/60
|
|
Refractive error
|
Mild myopic astigmatism
|
Mild mixed astigmatism
|
Moderate myopic astigmatism
|
Mild compound astigmatism
|
Mild myopia
|
|
Pupils’ reaction to light
|
Sluggish
|
Sluggish
|
Sluggish
|
Sluggish
|
Sluggish
|
|
Fundus
|
|
|
|
|
|
|
Disc pallor
|
+
|
+
|
+++
|
++
|
++
|
|
Vessel attenuation
|
++
|
+
|
+++
|
++
|
+++
|
|
Macula
|
Atrophic
|
Dull
|
Atrophic
|
Dull
|
Dull
|
|
Pigmentary changes in the retinal background
|
Not present
|
Not present
|
Mild granularity
|
Not present
|
Mild granularity
|
|
ERG
|
Severe cone-rod dysfunction
|
Severe cone-rod dysfunction
|
Severe cone-rod dysfunction
|
Severe cone - moderate rod dysfunction
|
Severe cone-rod dysfunction
|
|
Age when ERG done
|
2 years
|
2.5 years
|
2 years
|
7 years
|
25 years
|
|
Neurological features
|
|
Age of onset of symptoms
|
14 months
|
3 years 6 months
|
11 months
|
13–14 months
|
15yrs
|
|
Central hypotonia
|
+
|
+
|
+
|
NA
|
NA
|
|
Lower limbs Spasticity
|
+
|
-
|
+
|
+
|
+
|
|
Intention tremor
|
+
|
+
|
-
|
+
|
+
|
|
Cognitive disabilities
|
Mild intellectual disability
|
Not assessed
|
Learning difficulty; no formal assessment
|
Learning difficulty; no formal assessment
|
No
|
|
Overall motor function
|
Wheelchair-bound
|
Can walk with support
|
Pull up to stand and cruise around furniture
|
Able to walk with support
|
Wheelchair-bound
|
|
NCS
(age when detected)
|
normal
(6 years)
|
normal
(3 years 7 months)
|
not done
|
not done
|
mild motor neuropathy (25 years)
|
* Vision tested with sugar pearl test (single dark pearl identification is equivalent to Snellen 6/60).
SD: standard deviation; CSM: Central, Steady, Maintained; CUSM: central, steady, unmaintained; FF: Fixing and Following; US: Unsteady; OD: right eye, OS: left eye, OU: both eyes; ACBD5: Acyl-coenzyme A-binding domain-containing protein 5. Grading of findings: + mild, ++ moderate, +++ severe, - absent; ERG: Electroretinogram; NCS: nerve conduction studies; CS: corticospinal, MRI: magnetic resonance imaging; NA: not available.
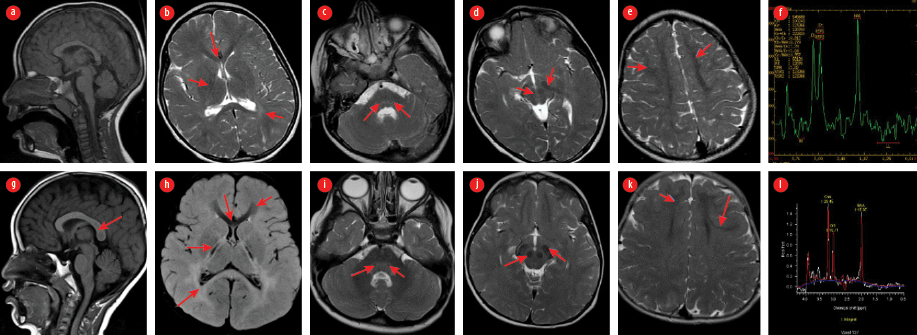 Figure 1: a–f: Comparative MRI brain of Patient 1 obtained at age of 4 years. MRI o f the brain of his brother (Patient 2) obtained at age of 3 years of the age: (a and g) sagittal T1W, (h) axial fluid-attenuated inversion recovery, (b, c, d, e, i, j, and k) axial SE T2W images and (f and l) MRI showing diffusely altered hypomyelinated/demyelinated periventricular and deep white matter with posterior pattern due to dominant involvement of parietal and (b and h) occipital lobes, (b, g, and h) splenium corpus callosum and (c and i) posterior pontine tegmentum; (e and k) visible residual preserved hypointense myelinated white matter at frontal lobes, also with sparing subcortical fibers and (b and h) genu of corpus callosum; consequent striking appearance of posterior limb of internal capsule, (d and j) substantia nigra and (c and i) red nuclei in midbrain and medial lemniscus identified as hypointense dots in posterior pontine tegmentum; (f) MRI spectroscopy correlates with metabolic disorder presenting in case one with moderately decreased N acetylaspartate and (l) increased choline and more significantly lower N acetylaspartate and elevated Choline in case two; all described structures are marked with arrows on mentioned images.
Figure 1: a–f: Comparative MRI brain of Patient 1 obtained at age of 4 years. MRI o f the brain of his brother (Patient 2) obtained at age of 3 years of the age: (a and g) sagittal T1W, (h) axial fluid-attenuated inversion recovery, (b, c, d, e, i, j, and k) axial SE T2W images and (f and l) MRI showing diffusely altered hypomyelinated/demyelinated periventricular and deep white matter with posterior pattern due to dominant involvement of parietal and (b and h) occipital lobes, (b, g, and h) splenium corpus callosum and (c and i) posterior pontine tegmentum; (e and k) visible residual preserved hypointense myelinated white matter at frontal lobes, also with sparing subcortical fibers and (b and h) genu of corpus callosum; consequent striking appearance of posterior limb of internal capsule, (d and j) substantia nigra and (c and i) red nuclei in midbrain and medial lemniscus identified as hypointense dots in posterior pontine tegmentum; (f) MRI spectroscopy correlates with metabolic disorder presenting in case one with moderately decreased N acetylaspartate and (l) increased choline and more significantly lower N acetylaspartate and elevated Choline in case two; all described structures are marked with arrows on mentioned images.
Case two (male, 4 years old)
The patient, the younger brother of the patient in Case one, developed nystagmus and photosensitivity at five months old. Although he had begun to walk independently at 15 months, by the age of three years his gait rapidly became unsteady, followed by a complete inability to walk unassisted. His speech progressed well during the first two years but regressed thereafter.
At the age of four, the child’s head circumference was at 10th percentile, weight at 8th percentile and height at 2nd percentile. Physical examination revealed head titubation with intention tremor, hypotonia with hyperextensible large joints, bilateral Achilles tendon contractures, and brisk deep tendon reflexes. Visual evoked potential yielded no response in the right eye and a normal response in the left eye. Ophthalmic examination showed nystagmus, photosensitivity, severe visual impairment, mild mixed astigmatism, pale discs, attenuated vessels, and a dull foveal reflex in both eyes. ERG showed severe cone-rod dysfunction. Brain MRI/ magnetic resonance scanning (MRS) features as detailed in Table 1 [Figure 1: g–l]. WES (Centogene, Rostock, Germany) revealed a homozygous deletion involving in exons 7 and 8 of the ACBD5 gene on copy number variation analysis.
Case three (female, 7 years old)
This patient was the second-born child of a first-cousin couple. Her history included nystagmus and photophobia at five months of age, and lower-limb spasticity was noted at 11 months, when she started to pull to stand. Brain MRI at three years of age showed bilateral signal abnormalities of deep white matter, mainly involving the posterior limb of the internal capsule and splenium.
At the current presentation (seven years old), all her growth parameters were below the second percentile with weight at -4.8 SD, height at -4.5 SD, and head circumference at -3 SD. She was cruising and crawling around furniture, but was unable to take independent steps. Despite clear speech, she showed significant learning disability. Neurological examination revealed upper limb hypertonia, severe lower limb spasticity, hyperreflexia, and extensor plantar response bilaterally. There was no intention tremor or dysarthria.
Ophthalmic examination was positive for nystagmus, photosensitivity, moderate myopic astigmatism, and severe visual impairment. Fundus examination revealed disc pallor, attenuated vessels, and mild granularity of the background retina with pigmentary changes at the macula, which progressed to an atrophic macula. Visual evoked potential showed significant delayed P2 latency and severely reduced P2 amplitude, indicating severe nerve fibre loss. ERG revealed severe cone-rod dysfunction. VLCFA profile showed elevated C24:0 at 84.25 umol/L (18.99–72.54) and C26:0 at 1.71 umol/L (0.00–1.08). However, the C26:0/C22 and C26:0/C24:0 ratios were normal, as was the level of phytanic acid. Retinal dystrophy gene panel (Manchester University, NHS Foundation Trust, Manchester, UK) revealed a homozygous deletion of exons 7 and 8 of the ACBD5 gene.
Case four (female, 10 years old)
The eldest offspring of double-first-cousin parents, this patient’s history included horizontal nystagmus and photophobia at the age of three months. She attained independent walking at 13 months of age, but by 2 years of age, she was found to have gait unsteadiness, intention tremors, and lower-limb hyperreflexia. By the age of three, her gait worsened with frequent falling and eventually progressed to limited independent walking and severely spastic gait. At age 10 years, she continued to have poor academic performance. Her weight, height, and head circumference ranged between the 5th and 10th percentiles. Neurological examination revealed intention tremors, lower limb spasticity, and generalized hyperreflexia with bilateral ankle clonus. Ophthalmic examination results included severe visual impairment, pale optic discs, attenuated retinal arterioles, dull foveal reflexes, and pigmentary retinopathy [Figure 2]. ERG confirmed severe cone-rod dystrophy.
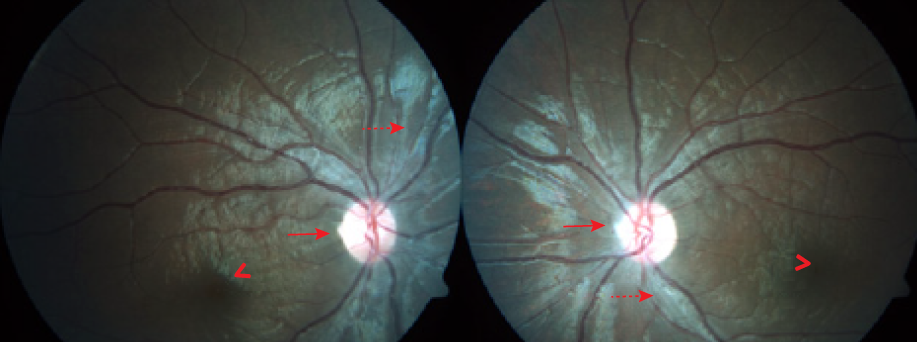 Figure 2: Case four fundus photographs showing moderate diffuse optic disc pallor (solid arrows), moderate arteriolar attenuation (dashed arrows), dull macular and foveal reflexes (arrow heads), and normal retinal background.
Figure 2: Case four fundus photographs showing moderate diffuse optic disc pallor (solid arrows), moderate arteriolar attenuation (dashed arrows), dull macular and foveal reflexes (arrow heads), and normal retinal background.
Brain MRI at the age of four years showed bilateral confluent white matter changes—mainly in the periventricular and splenium areas. MRS showed moderately decreased NAA and elevated Choline. VLCFA was mildly elevated with C26:0 at 1.24 umol/L (0.00–1.08) and C24:0/C22:0 ratio at 1.288 (0–1.158), with a normal pristanic acid level. WES (Breda Genetics, Brescia, Italy) identified a homozygous deletion in exon 4 of the ACBD5 gene.
Case five (male, 30 years old)
This patient was born to consanguineous parents. Photophobia and nystagmus were noted in the first months of his life, with progressive visual impairment, due to which he attended a school for children with visual needs. Gait unsteadiness was first noted at the age of 15 years. At 17, he had progressive walking difficulties and ataxia. He did not report any swallowing difficulties and has no dysarthria. There was no history of urine or bowel incontinence, seizures, memory impairment, or behavioral changes. Ophthalmology assessment at 21 showed nystagmus, severe visual impairment, disc pallor, attenuated vessels, and pigmentary changes at the macula. ERG confirmed severe cone-rod dysfunction. Nerve conduction study at the age of 25 years showed mild motor neuropathies (axonopathy). Brain MRIs at 17 and 25 years of age are detailed in Table 1 and Figure 3. WES (Centogene, Rostock, Germany) identified a homozygous deletion in exon 4 of the ACBD5 gene.
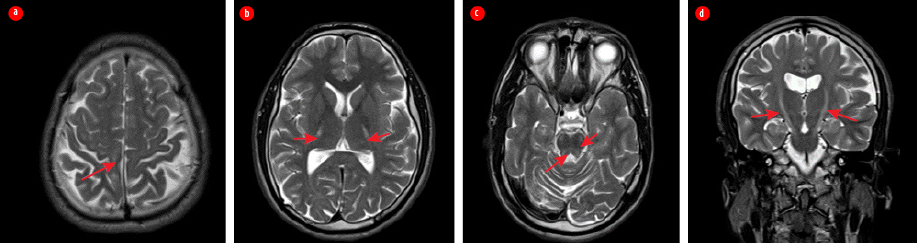 Figure 3: Case five MRI of the brain taken at 25 years of age. (a) Axial T2-weighted images showing hyperintensity of corticospinal tracts (arrows) bilaterally from perirolandic cortex (b) throughout posterior limb of internal capsule (c) reaching the pontine level; (c) visible atrophic changes of cerebellum and (a)parietal lobes on brain convexity; (c) also presenting signal alteration at posterior pontine tegmentum and both superior cerebellar peduncles. (d) Coronal T2W image showing bilateral involvement of the corticospinal tract.
Figure 3: Case five MRI of the brain taken at 25 years of age. (a) Axial T2-weighted images showing hyperintensity of corticospinal tracts (arrows) bilaterally from perirolandic cortex (b) throughout posterior limb of internal capsule (c) reaching the pontine level; (c) visible atrophic changes of cerebellum and (a)parietal lobes on brain convexity; (c) also presenting signal alteration at posterior pontine tegmentum and both superior cerebellar peduncles. (d) Coronal T2W image showing bilateral involvement of the corticospinal tract.
The patient is university-educated and currently employed as a medical recorder in a hospital. Physical examination showed a slim man with a body mass index of 19 and a head circumference at the second percentile. His sensations of temperature, pain, and touch were intact with an impaired sense of joint positions of the lower limbs, intention tremor, and dysmetria. He had normal muscle tone, hyperreflexia, and bilateral extensor plantar response. Muscle strength was normal in the upper limbs. However, lower limbs had bilateral pes cavus and significant distal weakness with no joint contractures. The patient was fully wheelchair-dependent.
Discussion
As explained earlier, the physiological importance of the ACBD5 gene in peroxisomal β-oxidation of VLCFAs was first discovered in 2013.5 The cardinal features of ACBD5-related retinal dystrophy with leukodystrophy (RDLKD) were consistent with subsequent reports and included infantile-onset nystagmus followed by motor deterioration, spasticity, cerebellar symptoms, and cognitive disability.3,6,7 Ferdinandusse et al,3 showed that the ACBD5 defect leads to the accumulation of VLCFAs due to their impaired peroxisomal β-oxidation. They demonstrated increased levels of C26:0 lysophosphatidylcholine (C26:0 lysoPC) and/or C26-acylcarnitine in plasma, dried blood spot, and fibroblasts from their patient with other peroxisomal parameters in blood (plasma phytanic acid, pristanic acid, pipecolic acid, and plasmalogens in erythrocytes) being normal, in a pattern that was similar to patients with X-linked adrenoleukodystrophy. The group also confirmed the absence of ACBD5 protein in the HeLa cells (HeLaΔACBD5) by immunoblotting.
The oldest patient reported to date, a 36-year-old Brazilian woman, who, in addition to being wheelchair-bound, had developed significant cognitive decline, upper extremity weakness, difficulty with fine motor movements, sphincter incompetence, and neurogenic bladder during her teens, and severe dysphagia with gastrostomy dependence in her thirties.6 However, the child from the UAE had additional malformations exhibited by facial dysmorphism and cleft palate. Interestingly, this patient was homozygous for the exon 7 and 8 deletion in the ACBD5 gene, which was also identified in three patients in our cohort. None of our five patients (including the three with the same variant as in the UAE patient), the Brazilian woman,6 or the Turkish sisters,7 had the extra features described by the Ferdinandusse group, supporting the assumption that the malformations observed in the UAE child were unlikely to be related to ACBD5 deficiency.
The neurological symptoms of all four children in the current series were evident by age three. Our adult patient (case five), however, had no motor limitations until he was fifteen. He is also our only patient with normal cognitive function at the age of 30. It is difficult to correlate the differences in his neurological function with his genotype, as he shares the same homozygous deletion of exon 4 with case four, whose neurological manifestations began when she was two years old.
Growth restriction and microcephaly were also frequent features among patients in the current series, specifically those wuth a homozygous deletion in exons 7 and 8; however, there was no growth abnormality or microcephaly in cases four and five, both of which harbor the same homozygous out-of-frame deletion of exon 4.
Retinopathy is a recurrent pathology in patients with disorders of peroxisome biogenesis. Research on rodent retina8 was able to detect peroxisomes in all retinal layers, with high expression of ACBD5 in retinal outer segments.9 The latter finding suggests that polyunsaturated fatty acids (PUFA) and VLCFA, besides being recycled to the photoreceptors, can be taken up and degraded by peroxisomes.9 It was also shown that ACBD5 functions to tether peroxisomes to the endoplasmic reticulum.10 Retinopathy in ACBD5 mutations is assumed to be related to impairment in these functions and impaired metabolism of VLCFA and PUFA, which are crucial components of the retina. Retinal dystrophy occurred in virtually all patients with ACBD5 null mutations reported to date, and their ocular manifestations were very similar in the age of onset, presenting symptoms, fundus findings, and ERG finding s consistent of cone-rod dystrophy. It seems that retinal involvement does not correlate with age of onset or severity of the neurological disease, suggesting that milder missense variants may manifest as isolated non-syndromic retinal dystrophy.
The VLCFA levels were assessed in three of our patients, who showed a similar pattern to the four previously reported patients with elevations of C26:0 with or without elevations of C24:0, C26:0/C22:0, and C24:0/C22:0 ratios. Although nonspecific, this biochemical parameter can provide an important clue in diagnosing this form of peroxisomal disorder in patients with consistent clinical findings.
The neuroradiological features of patients in this series are fairly consistent and show a recognizable pattern similar to previously reported patients with hypomyelination with diffuse hyperintense T2 and fluid-attenuated inversion recoverysignal abnormality in white matter with relative sparing of the subcortical U fibers that extend along the cerebrospinal tracks involving bilateral cerebral peduncles up to the brain stem. The demonstration of atrophic changes in the bilateral cerebellar hemispheres, brainstem, and the thoracic spinal cord in the oldest patient in this series is similar to the oldest reported patient6 and supports the progressive neurodegenerative nature postulated for this disease. The metabolites revealed by MRS were also consistent in all our five patients and in previously reported patients with decreased NAA and increased choline.
Conclusion
This report supports the previous observations that ACBD5-related RDLKD is a well-recognized neurodegenerative disease of peroxisomal fatty acids beta-oxidation with early onset cone-rod dystrophy and variable age of onset of neurological decline that is characterized by progressive spastic paraparesis, ataxia, and cognitive decline. RDLKD should be included in the differential diagnosis of patients with retinal dystrophy associated with progressive spastic paraparesis and cerebellar symptoms.
Disclosure
The authors declare no conflicts of interest. Informed consent was obtained from the adult patient and from the legal guardians of all pediatric patients.
Acknowledgments
We wish to acknowledge all patients and their families for their participation relevant data and giving consent. We also thank Ms. Asila Al Habsi, the Biochemical Genetics Specialist Nurse, for following up the patients and obtaining consents, and Dr. Muna Al Ruhaili, Ophthalmology resident, for her help in extracting patient information.
references
- 1. Waterham HR, Ferdinandusse S, Wanders RJ. Human disorders of peroxisome metabolism and biogenesis. Biochim Biophys Acta 2016 May;1863(5):922-933.
- 2. Wiese S, Gronemeyer T, Ofman R, Kunze M, Grou CP, Almeida JA, et al. Proteomics characterization of mouse kidney peroxisomes by tandem mass spectrometry and protein correlation profiling. Mol Cell Proteomics 2007 Dec;6(12):2045-2057.
- 3. Ferdinandusse S, Falkenberg KD, Koster J, Mooyer PA, Jones R, van Roermund CW, et al. ACBD5 deficiency causes a defect in peroxisomal very long-chain fatty acid metabolism. J Med Genet 2017 May;54(5):330-337.
- 4. Yagita Y, Shinohara K, Abe Y, Nakagawa K, Al-Owain M, Alkuraya FS, et al. Deficiency of a retinal dystrophy protein, Acyl-CoA binding domain-containing 5 (ACBD5), impairs peroxisomal β-oxidation of very-long-chain fatty acids. J Biol Chem 2017 Jan;292(2):691-705.
- 5. Abu-Safieh L, Alrashed M, Anazi S, Alkuraya H, Khan AO, Al-Owain M, et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res 2013 Feb;23(2):236-247.
- 6. Bartlett M, Nasiri N, Pressman R, Bademci G, Forghani I. First reported adult patient with retinal dystrophy and leukodystrophy caused by a novel ACBD5 variant: A case report and review of literature. Am J Med Genet A 2021 Apr;185(4):1236-1241.
- 7. Gorukmez O, Havalı C, Gorukmez O, Dorum S. Newly defined peroxisomal disease with novel ACBD5 mutation. J Pediatr Endocrinol Metab 2021 Oct;35(1):11-18.
- 8. Das Y, Roose N, De Groef L, Fransen M, Moons L, Van Veldhoven PP, et al. Differential distribution of peroxisomal proteins points to specific roles of peroxisomes in the murine retina. Mol Cell Biochem 2019 Jun;456(1-2):53-62.
- 9. Bazan NG. Cellular and molecular events mediated by docosahexaenoic acid-derived neuroprotectin D1 signaling in photoreceptor cell survival and brain protection. Prostaglandins Leukot Essent Fatty Acids 2009;81(2-3):205-211.
- 10. Hua R, Cheng D, Coyaud É, Freeman S, Di Pietro E, Wang Y, et al. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J Cell Biol 2017 Feb;216(2):367-377.