|
Abstract
Hypocomplementemic urticarial vasculitis syndrome is an immune complex-mediated disease of unknown etiology. The clinical course is characterized by urticaria, conjunctivitis, joint pain, and hypocomplementemia. We here report a case of a child with hypocomplementemic urticarial vasculitis syndrome that progressed to nephritis. Renal biopsy was consistent with diffuse proliferative glomerulonephritis with diffuse subendothelial immune deposits. He responded well to a combination of steroid and mofetil micofenolate.
Keywords: HUVS; Nephritis; Children.
Introduction
Hypocomplementemic urticarial vasculitis syndrome (HUVS) is a relatively uncommon disease which has mainly been reported in the fourth decade of life. HUVS consists of recurrent urticarial vasculitis, arthralgia/arthritis and low complements. Angioedema, ocular inflammation, glomeulonephritis and obstructive lung disease are other clinical findings that have been observed in this syndrome.1 HUVS is also reported in children with a common presentation of episodes of urticaria, eye inflammation and hypocomplementemia.2 Here, we report of a child with HUVS and nephritis that required immunosuppressive treatment.
Case Report
An 8-year-old boy presented at the age of 3 years with episodes of urticarial rashes, red eyes, knee pain and low-grade fever for 4 months. Two weeks prior to presentation, he had swelling in the cervical region. On examination, he looked short and asthenic. His weight and height were 12.9 kg (25 centile) and 114 cm (25 centile), respectively. The systems examination revealed injected conjunctive, generalized lymphadenopathy, cervical lymph nodes measuring 2 × 2 cm, with no hepato or splenomegally and wheals scattered in the trunk, upper and lower extremities. (Fig. 1)
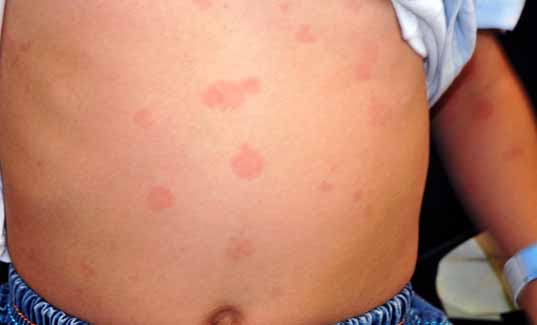
Figure 1: Urticarial rash.
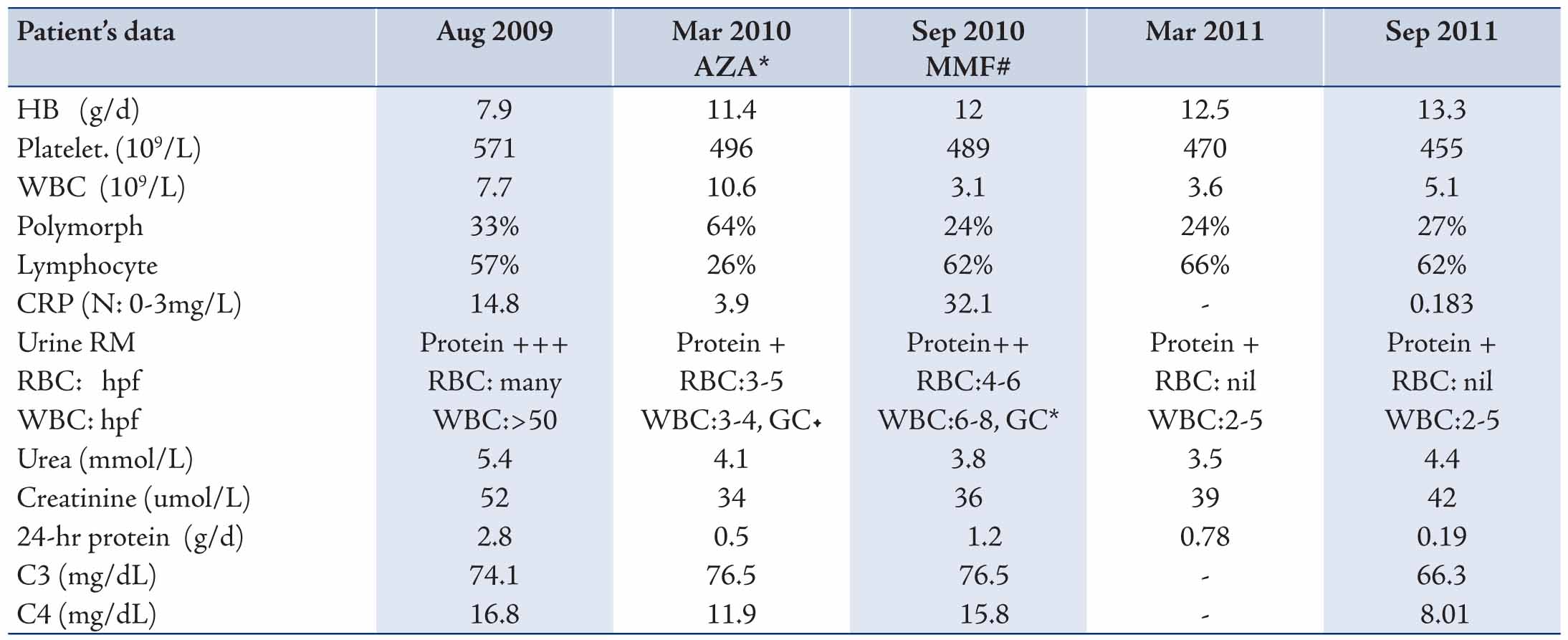
Laboratory investigations showed the following: The complete blood count (CBC), liver function test (LFT), serum urea, creatinine, electrolytes (U/E/Cr), ESR and C- reactive protein (CRP) were all unremarkable. The serology screening for infections revealed Epstein Barr Virus (EBV) IgM and IgG positive. Rheumatology workup was negative for antinuclear antibody (ANA), extractable nuclear antigen antibody (ENA), or antistreptolysion O (ASO) titre; however, his plasma complement levels for C3 was low at 74 mg/dL (N: 90-180) and C4 was normal. Urine analysis was initially normal. During a follow-up visit 2 months later, there were no changes in the size of the cervical lymph nodes, for which lymph node biopsy was done which gave a picture of reactive lymphadenitis. Repeated EBV serology was positive for IgG and EBNA.
In the following 2 years, he had recurrent episodes of urticarial rashes and conjunctivitis noticed every 3-4 weeks; each episode lasted for 2-3 days and improved with oral antihistamines. Ophthalmology review reported only conjunctivitis. He was gaining weight and height but remained in between the 10th and 25th centiles. Repeated laboratory tests including urinalysis were normal. Further serology workup including antineutrophil cytoplasmic antibody (ANCA), anticardiolipin antibody and anticyclic citrulinated peptide (CCP) antibody were also negative. HLA testing revealed class 1 A2, A24, B61 and B70. Serum immunoglobulins were normal but C3 was consistently low. C2 complement was normal while CH50 was low. C1 inhibitor was 0.6 g/L (0.18-0.33). Cryoglobulin was negative, and protein electrophoresis was normal.
Three years after the first presentation and in spite of being clinically free except for the urticarial rashes, he was noticed to have abnormal urinalysis that showed microscopic hematuria (RBC’s), pyuria (WBC’s), proteinuria and granular casts which were seen occasionally. The 24-hour proteinuria was 2.8 g/day with normal renal function. He was also anemic with high inflammatory markers. (Table 1)
Skin biopsy was performed on the new urticarial rashes, and it was reported to be leukocytoclastic vasculitis. A percutaneous kidney biopsy was done and revealed features of a diffuse proliferative glomerulonephritis with crescent formations. Interstitium showed edema and focal lymphocytic infiltrate. The blood vessels showed hyaline arteriolosclerosis. Immunofluorescence staining showed blood vessel deposits of IgA (++), IgM (+), IgG (+++), C3 (+++), CIq (++) and fibrinogen (+), which was suggestive of immune-complex vasculitis. He was treated with IV pulses of methylprednisolone followed by oral prednisolone 2 mg/kg/day.
Later, he developed remarkable redness in his eyes diagnosed as episcleritis. However, the episcleritis spontaneously improved and 4 weeks later, he went into a remission, where the urinalysis became normal. His 24-hour urine protein also became normal, and ESR was below 20 mm/hr. C3 levels increased to 100 mg/dL (90-180). He had no further attacks of eyes or skin problems; however, after tapering the prednisolone dose, he had variant amount of proteinuria in the following few months. His urinalysis again showed pyuria and hematuria. C3 increased but to below normal value. Repeated serology screenings for ANA and ANCA were negative. At this time, anti C1q antibody was requested and the results were negative. (Table 1)
Because of persistent proteinuria, the patient had a second renal biopsy 8 months later. The biopsy revealed 6 glomeruli, 3 of which were globally sclerosed and the other 3 showed diffuse mesengial cell hyperplasia. The conclusion was most probably resolving glomerulonephritis with mild focal tubular atrophy, (Fig. 2). At this stage, azathioprine (AZA) was added at a dose of 1.5 mg/kg/day.
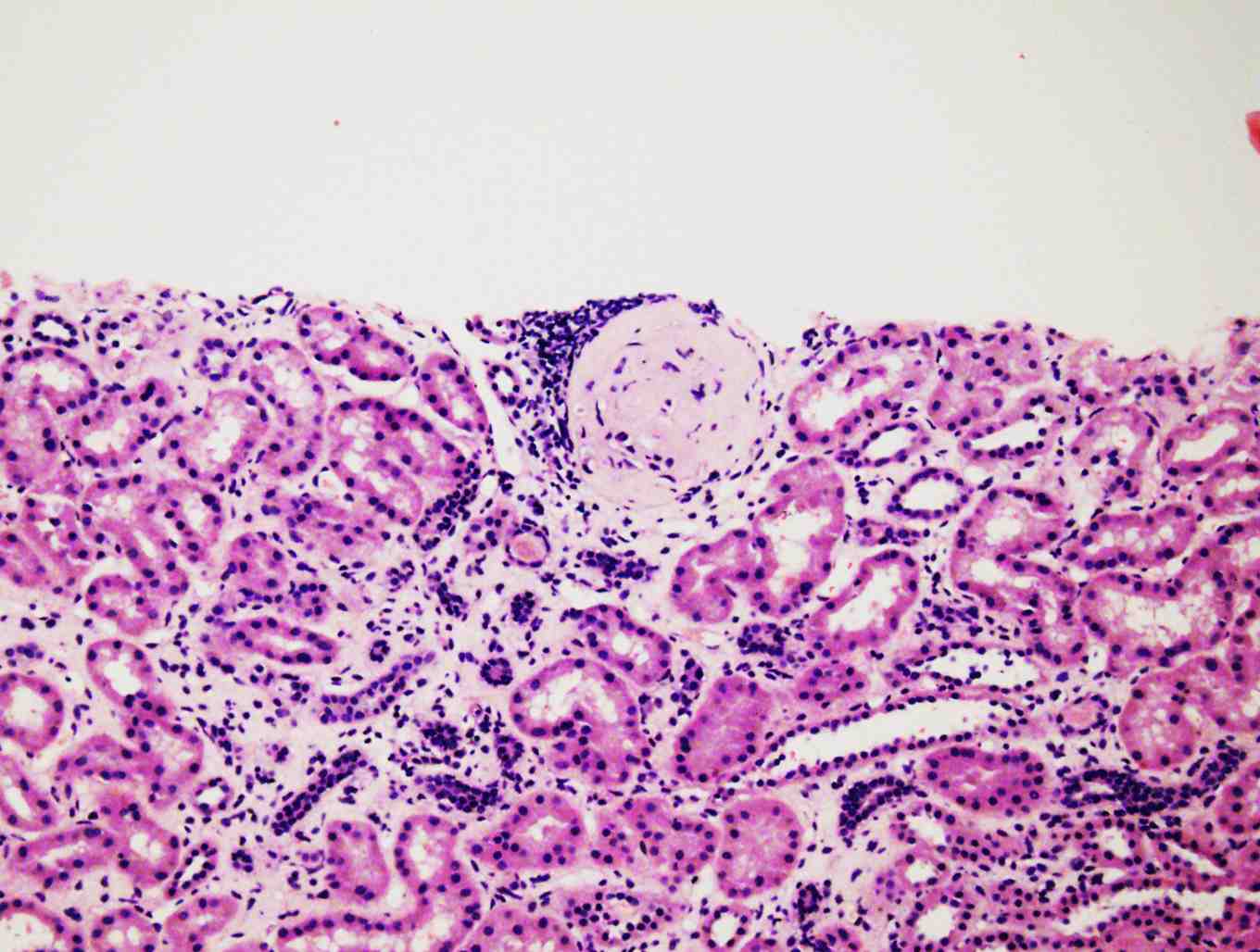
Figure 2: Diffuse mesangial cell hyperplasia and sclerosed glomeruli.
Two months later, he had a flare-up in the eyes, skin, and the kidney nephritis on tapering of the prednisolone dose to 0.5 mg/kg every other day. Therefore, he was managed with 2 pulses of methylprednisolone followed by increasing the dose of prednisolone. On the second trial of decreasing the prednisolone dose the, patient had a flare-up in skin and kidney and azathioprine was changed to mofetil micofenolate (MMF). Six months later, his urinalysis revealed protein +, WBC 2-5/ hpf, and no RBC. His 24-hour urine protein was 0.78 g/L. (Table 1)
At age of 7½ years, he looked short and asthenic with few residual urticarial rashes. His weight was 22.9 kgs (10th centile) and height 116 cm (3rd centile). However, his school performance was reported as being well.
Table 1: The laboratory results of the patient during the course of illness.
Discussion
Hypocomplementemic urticarial vasculitis syndrome (HUVS) is a syndrome of recurrent urticarial vasculitis, arthralgia/arthritis, and hypocomplementemia which was first described by McDuffie et al. in 1973.1 It is a rare entity which is well described in adults but has also been observed in children.2 HUVS is a specific autoimmune disease with involvement of the skin, joints, kidneys and gastrointestinal tract manifested by vasculitis and polyserositis. Occasionally, other organs are involved such as the eyes, lungs and nervous system.3
Antibodies against C1q act as a diagnostic marker for HUVS. However, they are not specific to HUVS since they have also been observed in SLE.4 Direct immunofluorescence (DIF) of skin biopsy samples from HUVS patients usually shows immunoglobulin and complement activation in a granular pattern in and around the blood vessels of the upper dermis and the basement membrane which is similar to skin biopsy findings of SLE patients.5 C1q immune complexes were found in the kidney biopsy of our patient, but we could not detect anti-C1q antibody in the serum which may be attributed to using steroid therapy prior to the blood test.
Some authors have suggested the possible progression of HUVS to SLE and others have considered it as a minor form of SLE. Serum ANA is positive in 50% of patients with HUVS while anti-dsDNA antibody and ENA are rarely seen.6
In MEDLINE review of HUVS literatures, we were not able to find concurrence of HUVS and acute EBV infection that our patient presented with on his first visit. However, there was a 3-month interval between the HUVS episodes and the appearance of lymph node enlargement. We may hypothesize that deregulated immune status in HUVS would predispose to such infection.
Table 2: Summary of different published studies about HUVS manifestations in children.
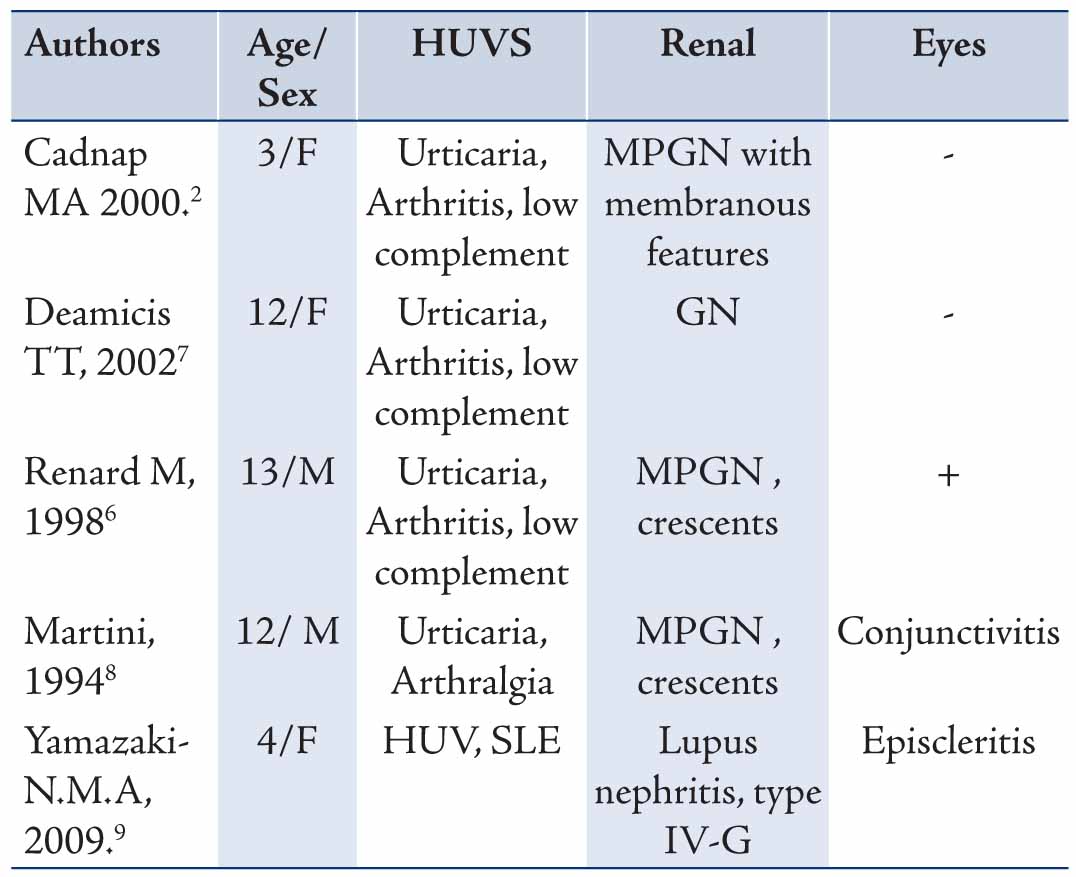
Glomerulonephritis (GN) can develop after many years of HUVS symptoms and can be rapidly progressive.7 Despite that, our patient had recurrent episodes of skin rash and eye redness for more than 3 years, he had no renal manifestation. He only developed abnormal urinalysis 3 years following the initial presentation. In MEDLINE search for children with HUVS, all reported cases, except one with SLE, had nephritis without SLE serological features, (Table 2). Two of these cases (cases 2 and 4) behaved clinically like our patient and had renal complications many years after skin and eye manifestations.
There is no consensus on the appropriate treatment of HUVS in the literature. Antihistamines are used for the skin lesions, while nonsteroidal anti-inflammatory drugs are used for arthralgia. Corticosteroids are used for skin manifestations and glomerulonephritis. Other immunosuppressive drugs including azathioprine, methotrexate and cyclophosphamide were considered for refractory HUVS.10,11 Since our patient discloses good response to mofetil micofenolate (MMF) in combination with steroid, we may defer the other immune suppressive drugs for the future.
Conclusions
We reported a case of an 8-year-old boy with HUV syndrome and diffuse proliferative glomerulonephritis that revealed good response to a combination of steroid and mofetil micofenolate (MMF) therapy.
Acknowledgements
The authors reported no conflict of interest and no funding was received for this work.
References
1. McDuffie FC, Sams WM Jr, Maldonado JE, Andreini PH, Conn DL, Samayoa EA. Hypocomplementemia with cutaneous vasculitis and arthritis. Possible immune complex syndrome. Mayo Clin Proc 1973 May;48(5):340-348.
2. Cadnapaphornchai MA, Saulsbury FT, Norwood VF. Hypocomplementemic urticarial vasculitis: report of a pediatric case. Pediatr Nephrol 2000 Apr;14(4):328-331.
3. Grotz W, Baba HA, Becker JU, Baumgärtel MW. Hypocomplementemic urticarial vasculitis syndrome: an interdisciplinary challenge. Dtsch Arztebl Int 2009 Nov;106(46):756-763.
4. Wisnieski JJ, Jones SM. IgG autoantibody to the collagen-like region of Clq in hypocomplementemic urticarial vasculitis syndrome, systemic lupus erythematosus, and 6 other musculoskeletal or rheumatic diseases. J Rheumatol 1992 Jun;19(6):884-888.
5. Mehregan DR, Hall MJ, Gibson LE. Urticarial vasculitis: a histopathologic and clinical review of 72 cases. J Am Acad Dermatol 1992 Mar;26(3 Pt 2):441-448.
6. DeAmicis T, Mofid MZ, Cohen B, Nousari HC. Hypocomplementemic urticarial vasculitis: report of a 12-year-old girl with systemic lupus erythematosus. J Am Acad Dermatol 2002 Nov;47(5)(Suppl):S273-S274.
7. Renard M, Wouters C, Proesmans W. Rapidly progressive glomerulonephritis in a boy with hypocomplementaemic urticarial vasculitis. Eur J Pediatr 1998 Mar;157(3):243-245.
8. Martini A, Ravelli A, Albani S, De Benedetti F, Massa M, Wisnieski JJ. Hypocomplementemic urticarial vasculitis syndrome with severe systemic manifestations. J Pediatr 1994 May;124(5 Pt 1):742-744.
9. Yamazaki-Nakashimada MA, Duran-McKinster C, Ramírez-Vargas N, Hernandez-Bautista V. Intravenous immunoglobulin therapy for hypocomplementemic urticarial vasculitis associated with systemic lupus erythematosus in a child. Pediatr Dermatol 2009 Jul-Aug;26(4):445-447.
10. Soter NA. Urticarial venulitis. Dermatol Ther 2000;13:400-408 .
11. Berg RE, Kantor GR, Bergfeld WF. Urticarial vasculitis. Int J Dermatol 1988 Sep;27(7):468-472.
|