Type 1 diabetes mellitus (T1DM) is a genetically heterogeneous autoimmune disease affecting about 0.3% of Caucasian populations.1 Genetic studies of T1DM have focused on the detection of loci associated with increased susceptibility to this multifactorial phenotype.1 Many patients eventually develop diabetic ketoacidosis, which can be a major risk of mortality.
Polycystic kidney disease (PKD) is one of the most common hereditary kidney diseases affecting the renal tubules. The affected individuals may carry autosomal dominant PKD (ADPKD) and autosomal recessive PKD (ARPKD). ADPKD has an estimated prevalence of 1:400 to 1:1000 live births.2,3
Genetically, ADPKD is a heterogeneous disease mainly caused by mutations in PKD1 and PKD2 genes, although 5–10% of ADPKD pedigrees remain either genetically unsolved or harbor rare mutations in other genes causing ADPKD-like phenotypes such as α-glucosidase neutral AB, DNAJB11, or hepatocyte nuclear factor 1β (HNF1B) gene.4
Case report
A 35-year-old man, with a known case of metabolic syndrome and a body mass index of 30 Kg/m2, had hypertension since early adolescence and T1DM since the age of four years. His diabetes was being maintained on insulin, dyslipidemia on statin, and hyperuricemia on medications. He also had chronic kidney disease – secondary to his ADPKD. He developed end-stage kidney disease (ESKD) by the age of 32 years for which he underwent preemptive simultaneous pancreatic and kidney transplant from a deceased donor. He was discharged with a serum creatinine of 78 umol/L, estimated glomerular filtration rate > 90 mL/min/1.73 m2, and suboptimal blood sugar of 10 mmol/L.
Two days later, the patient was readmitted with 38.8 °C fever, chills, generalized weakness, fatigue, and mild abdominal pain mainly around the umbilicus. He had no gastrointestinal symptoms, urinary tract symptoms, or other systemic symptoms. On examination, he was dehydrated, with blood pressure of 90/50 mmHg, pulse rate of 120/min, and respiratory rate of 22/min. Abdominal examination revealed mild distension and a well-healed scar was visible extending from the umbilicus to the suprapubic region. There was mild tenderness below the umbilicus. On palpation, the kidney graft was non-tender. Laboratory investigation results at admission are shown in Table 1.
While the patient’s blood cultures revealed no growth, his urine culture showed Enterococcus faecium, which was sensitive to vancomycin. The pus discharge from his surgical wound grew carbapenem-resistant Klebsiella pneumoniae ssp. (sensitive to tigecycline) and Citrobacter freundii (sensitive to cotrimoxazole). His chest X-ray revealed normal lung fields with prominent heart size. Abdominal X-ray revealed no abnormally, dilated bowel loops, or signs of free gas. Echocardiography showed mild pericardial effusion measuring 7 mm behind the right atrium and grade 1 diastolic dysfunction with an ejection fraction of 55%.
Table 1: Laboratory investigations on admission, after intervention drainage, and discharge.
|
Hemoglobin, g/dL
|
11.5–15.5
|
11.0
|
11.1
|
10.0
|
|
Total WBC, 10 g/dL
|
2.2–10.0
|
7.4
|
4.5
|
7.9
|
|
Neutrophils, 10 g/dL
|
1–5
|
6.4
|
3.3
|
6.2
|
|
Platelets, 10 g/dL
|
140–400
|
204
|
292
|
387
|
|
Lymphocytes, 10 g/dL
|
0.5
|
0.6
|
1.0
|
1.3
|
|
Blood urea, mmol/L
|
2.5–6.7
|
5.6
|
12.3
|
11.7
|
|
Serum creatinine, umol/L
|
45–100
|
83
|
88
|
136
|
|
Serum sodium, mmol/L
|
135–145
|
137
|
133
|
137
|
|
Serum potassium, mmol/L
|
3.5–5.0
|
5.1
|
4.3
|
4.9
|
|
eGFR, mL/min/1.73 m2
|
> 90
|
> 90
|
> 90
|
56
|
|
Serum insulin, pmol/L
|
20
|
227.3
|
64.3
|
64.3
|
|
Serum glucose, mmol/L
|
4.0–5.5
|
7.0
|
4.4
|
4.5
|
|
HbA1c, mmol/mol
|
20–42
|
37
|
35
|
39
|
|
Lipase-1, U/L
|
13–60
|
226
|
226
|
74
|
|
Amylase in serum, IU/L
|
8–55
|
127
|
111
|
130
|
|
C-peptide, pmol/L
|
260–1710
|
2363
|
716
|
2994
|
|
Serum albumin, g/L
|
35–50
|
23
|
28
|
30
|
|
ALT, IU/L
|
0–40
|
12
|
14
|
18
|
|
Anti-islet Ab
|
Cut off > 1.0 is positive
|
> 20
|
–
|
–
|
|
C-Reactive protein, mg/L
|
<5
|
14.2
|
1.4
|
4.4
|
|
ESR, mm/hr
|
2–25
|
39
|
22
|
-
|
|
CMV PCR, copies/mL
|
Nil
|
55 247
|
< 100
|
< 100
|
WBC: white blood cells; eGFR: estimated glomerular filtration rate; HbA1c: hemoglobin A1c; ALT: alanine transaminase; ESR: erythrocyte sedimentation rate; PCP: polymerase chain reaction.
Figure 1 shows the patient’s abdominal magnetic resonance imaging, with descriptions underneath.

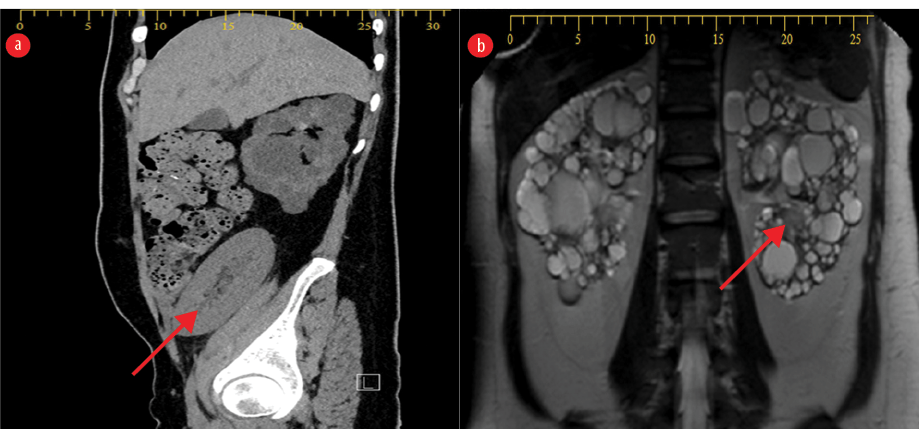 Figure 1: Abdominal MRI post kidney and pancreatic transplant. (a) The native kidneys show features of polycystic kidney disease, with innumerable variably sized renal cysts, replacing the normal renal parenchyma. The native pancreas is atrophied. The transplanted kidney is in the right iliac fossa (red arrow). (b) MRI of the abdomen shows multiple collections at the site of the transplanted kidney and pancreas, suggestive of abscess, likely infected hematomas. One of the collections is a large abdominopelvic, insinuating between the sigmoid and the transplanted pancreas (red arrow).
Figure 1: Abdominal MRI post kidney and pancreatic transplant. (a) The native kidneys show features of polycystic kidney disease, with innumerable variably sized renal cysts, replacing the normal renal parenchyma. The native pancreas is atrophied. The transplanted kidney is in the right iliac fossa (red arrow). (b) MRI of the abdomen shows multiple collections at the site of the transplanted kidney and pancreas, suggestive of abscess, likely infected hematomas. One of the collections is a large abdominopelvic, insinuating between the sigmoid and the transplanted pancreas (red arrow).

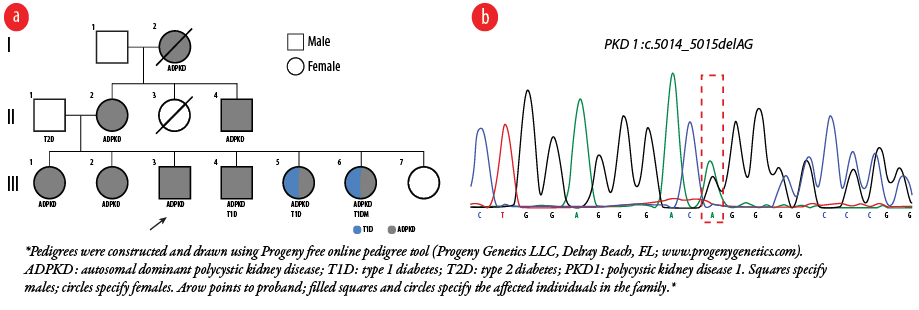 Figure 2: Genetic evaluation diagram of the patient’s family. (a) Pedigrees of the family showing the distribution of ADPKD, T1D and T2D among the members. (b) Genomic DNA Sanger chromatograms show heterozygous PKD1 c.5014_5015delAG; p.Arg1672fs*98 frameshift deletion.
Figure 2: Genetic evaluation diagram of the patient’s family. (a) Pedigrees of the family showing the distribution of ADPKD, T1D and T2D among the members. (b) Genomic DNA Sanger chromatograms show heterozygous PKD1 c.5014_5015delAG; p.Arg1672fs*98 frameshift deletion.
Our multidisciplinary team decided on interventional drainage of the collection with a pigtail catheter which yielded 30 mL of frank thick pus, whose culture yielded K. pneumoniae with sensitivity to cotrimoxazole and tigecycline. Ultrasound image post drainage showed the transplanted kidney (12 cm) with normal echotexture, no hydronephrosis, resistivity index of 0.54–0.58, and with 6 mL peri-graft collection. Abdominal computed tomography showed a significant reduction in the pre-drainage pelvic collection.
The pigtail catheter was removed after a week of no further drainage and a repeat ultrasound showed peri-graft collection of only 6 mL. The patient was managed with a full course of intravenous antibiotics for three weeks. A month later, with his laboratory investigation results showing progressive improvement [Table 1], he was discharged on a small dose of insulin of 8 U/day, steroids tapered down to 2.5 mg once daily (OD). He was kept on tacrolimus 3 mg twice daily (BID), mycophenolate mofetil (CellCept) 1 g BID, and valganciclovir 900 mg OD. He continued to receive intervention tigecycline daily at our daycare facility.
Our patient had a strong family history of both ADPKD and DM. His father was diabetic without kidney disease and his mother had ADPKD without diabetes and underwent kidney transplant. One brother and two sisters of the patient had ADPKD; the brother had a kidney transplant. Two other sisters had ADPKD with T1DM and were on insulin. A younger sister had neither ADPKD nor diabetes at the time of assessment [Figure 2a].
The proband’s sister III-1 underwent targeted next-generation sequencing panel that contains polycystic kidney disease-associated genes (including PKD1, PKD2, PKHD1, HNF1B, REN, UMOD, and MUC), which revealed a heterozygous 2-bp deletion (NM_001009944.2; c.5014_5015delAG) in the coding region, exon 15, of the PKD1 gene. This mutation was then verified by Sanger sequencing [Figure 2b]. It was also revealed that our proband, his mother, and three sisters (III-2, III-5, and III-6) were all carriers.
Discussion
Inherited and congenital kidney disease is an essential cause of ESKD in the Omani population which is characterized by high rate of consanguinity (56.3%).5,6 Omani patients with inherited kidney disease start renal replacement therapy at a relatively young mean age of 29 years.7,8
Our patient, who had a strong family history of both T1DM and ADPKD, developed both these conditions in early childhood (caused by a heterozygous frameshift deletion mutation in PKD1) leading to early kidney failure. Enlarged kidneys with multiple bilateral cysts along with hepatic cysts and hypertension were the typical characteristics of ADPKD in his family for multiple generations. Our patient received a preemptive simultaneous pancreatic and kidney transplant from a deceased donor but multiple complications followed.
Both clinical investigations and molecular genetic analysis proved the diagnosis of ADPKD. He was found to carry a previously reported heterozygous 2-bp deletion (NM_001009944.2; c.5014_5015delAG) in exon 15 of the PKD1 gene.5 Mutations in PKD1 (16p13.3) account for around 85% of ADPKD patients and are expected to lead to ESKD by on average age of 53.3 years, much earlier than the average age of ESKD in patients with PKD2 (4q22.1) mutations (72.7 years).9 Furthermore, truncating PKD1 mutations are linked with ESKD onset at younger ages than non-truncating mutations.10 In agreement with these studies, our patient and several members of his family developed ESKD at younger ages (proband: 32 years, III-5: 35 years, and III-6: 34 years). Our patient’s family history confirms a possible association between the mutation types and phenotypic outcome. Hence, the age of onset of renal failure can be influenced by the causal mutation, highlighting the importance of molecular genetic analysis for ADPKD patients.
Regarding T1DM, genetic susceptibility, and environmental factors interact to form the basic element in its progression.11 In the pedigree of our report, T1DM in combination with ADPKD was diagnosed in our patient and his two sisters (III-5 and III-6) who were all found positive for glutamic acid decarboxylase and anti-islet cell antibodies. It is rare for three or more siblings to develop T1DM, even though such cases have been reported in large families and in other populations.12–14 The lifetime risk of developing T1D is increased in relatives depending on which human leukocyte antigen haplotypes are shared.15,16 The lifetime risk in siblings of T1D-affected patients is unidentified, and our report of the family of our patient should inspire clinicians to evaluate their risks of diabetes.
Conclusion
We have described an Omani patient diagnosed with early-onset T1D and with a heterozygous frameshift mutation in the PKD1 gene who developed ESKD in his early thirties. His family had a long history of ADPKD and T1D. The study of familial clustering of specific diseases is an essential concept in genetic epidemiology that facilitates lifetime risk evaluation and early assessment, both in patients and their relatives. We recommend early genetic diagnosis in such cases to facilitate early treatment and thereby prevent the disease progression and the
associated morbidity.
Disclosure
The authors declared no conflicts of interest. The study was approved by the Research and Ethical Review and Approval Committee of the Ministry of Health (MOH) in Oman (MH/DGP/R&S/PROPOSAL_APPROVED/18) and conducted per the Declaration of Helsinki. Informed consent was obtained from the patient and his family members.
Acknowledgments
We thank our patient and his family for their trust in us. Each person listed as an author has participated sufficiently in the intellectual content, the analysis of data, and/or the writing of the manuscript to take public responsibility for it. Each author has reviewed the manuscript, believes it represents valid work, and approves it for submission.
references
- 1. Todd JA, Aitman TJ, Cornall RJ, Ghosh S, Hall JR, Hearne CM, et al. Genetic analysis of autoimmune type 1 diabetes mellitus in mice. 1991. J Immunol 2014 Jul;193(1):7-12.
- 2. Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJ, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers 2018 Dec;4(1):50-50.
- 3. Stel VS, Awadhpersad R, Pippias M, Ferrer-Alamar M, Finne P, Fraser SD, et al. International comparison of trends in patients commencing renal replacement therapy by primary renal disease. Nephrology (Carlton) 2019 Oct;24(10):1064-1076.
- 4. Bergmann C, von Bothmer J, Ortiz Brüchle N, Venghaus A, Frank V, Fehrenbach H, et al. Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol 2011 Nov;22(11):2047-2056.
- 5. Al Alawi I, Al Salmi I, Al Rahbi F, Al Riyami M, Al Kalbani N, Al Ghaithi B, et al. Molecular genetic diagnosis of Omani patients with inherited cystic kidney disease. Kidney Int Rep 2019 Aug;4(12):1751-1759.
- 6. Al Alawi I, Molinari E, Al Salmi I, Al Rahbi F, Al Mawali A, Sayer JA. Clinical and genetic characteristics of autosomal recessive polycystic kidney disease in Oman. BMC Nephrol 2020 Aug;21(1):347.
- 7. Al Alawi I, Al Salmi I, Al Mawali A, Al Maimani Y, Sayer JA. End-stage kidney failure in Oman: an analysis of registry data with an emphasis on congenital and inherited renal diseases. Int J Nephrol 2017;2017:6403985.
- 8. Al Alawi IH, Al Salmi I, Al Mawali A, Sayer JA. Kidney disease in Oman: a view of the current and future landscapes. Iran J Kidney Dis 2017 Jul;11(4):263-270.
- 9. Torra R, Badenas C, Darnell A, Nicolau C, Volpini V, Revert L, et al. Linkage, clinical features, and prognosis of autosomal dominant polycystic kidney disease types 1 and 2. J Am Soc Nephrol 1996 Oct;7(10):2142-2151.
- 10. Harris PC, Hopp K. The mutation, a key determinant of phenotype in ADPKD. J Am Soc Nephrol 2013 May;24(6):868-870.
- 11. Steck AK, Rewers MJ. Genetics of type 1 diabetes. Clin Chem 2011 Feb;57(2):176-185.
- 12. Olamoyegun MA, Ala OA. Type 1 diabetes in a Nigerian family - occurrence in three out of four siblings: a case report. World J Diabetes 2019 Oct;10(10):511-516.
- 13. Kishi A, Kawabata Y, Ugi S, Iwai T, Tanaka Y, Yoshizaki T, et al. The onset of diabetes in three out of four sisters: a Japanese family with type 1 diabetes. A case report. Endocr J 2009;56(6):767-772.
- 14. Verge CF, Howard NJ, Rowley MJ, Mackay IR, Zimmet PZ, Egan M, et al. Anti-glutamate decarboxylase and other antibodies at the onset of childhood IDDM: a population-based study. Diabetologia 1994 Nov;37(11):1113-1120.
- 15. Pociot F, McDermott MF. Genetics of type 1 diabetes mellitus. Genes Immun 2002 Aug;3(5):235-249.
- 16. Redondo MJ, Eisenbarth GS. Genetic control of autoimmunity in type I diabetes and associated disorders. Diabetologia 2002 May;45(5):605-622.