Methemoglobinemia is a hereditary or acquired blood condition, characterized by the oxidation of iron in hemoglobin (Hb) from ferrous (Fe2+) to ferric (Fe3+) state, thereby losing its functional capacity to bind to oxygen in the lungs. Hereditary methemoglobinemia is a rare condition caused by deficiency in the enzyme cytochrome b5 reductase, whose function is to reduce methemoglobin (meth-Hb) to normal Hb with the help of nicotinamide adenine dinucleotide + hydrogen.1–3 Dyshemoglobin disorders can be symptomatic or asymptomatic.
Hb M disease, the congenital form of methemoglobinemia, is associated with autosomal dominant inheritance, where tyrosine amino acid is replaced for histidine in the globin chain of Hb causing decreased oxygen binding capacity.4 The meth-Hb levels will be < 30% and present with central cyanosis. Acquired methemoglobinemia is much more dangerous and can be life-threatening, leading to hypoxia, cardiac dysfunction, and mortality. Intravenous methylene blue, ascorbic acid, and dextrose are commonly used for treatment.3
Case report
A 12-year-old girl presented with fever and cough for five days with mild exercise intolerance. She had no paroxysmal nocturnal dyspnea, orthopnea, pedal edema, or decreased urine output. Clinical examination showed central cyanosis with four limb oxygen saturation of 83–85%, normal blood pressure, and grade 3/6 systolic murmur in the left second intercostal space with wide-fixed split-second heart sound. She was diagnosed with COVID-19 and a large atrial septal defect (ASD) of 14 mm with left to right shift with mild pulmonary arterial hypertension. Atrial blood gas analysis showed normal partial pressure of oxygen and partial pressure of carbon dioxide. Complete blood count revealed Hb of 10 g/dL with normal indices, total leucocyte count of 7000/mm3, and platelets 200×109/L. The patient’s COVID-19 was managed conservatively and resolved in 10 days. However, post-COVID, despite supplemental oxygen, the patient’s oxygen saturation did not improve which led us to consider a blood chemistry disorder.
The patient’s family history revealed that her mother had low oxygen saturation and had been operated on for symptomatic ASD at the age of 30, but her father and sister had normal oxygen saturation. With this clinical picture, we suspected abnormal Hb along with familial congenital heart disease. The blood samples of the patient and her mother gave us the diagnosis of abnormal Hb indicated by the blood sample turning chocolate brown on exposure to 100% oxygen [Figure 1]. This was further confirmed spectrophotometrically, which showed a meth-Hb level of 22% with a total meth-Hb concentration of 2.2 g/dL, leading to a diagnosis of methemoglobinemia. We started her on oral ascorbic acid 1500 mg/day. On follow-up, the meth-Hb level had fallen significantly to < 5% (normal: 1–3%). She was operated on for ASD and discharged successfully.

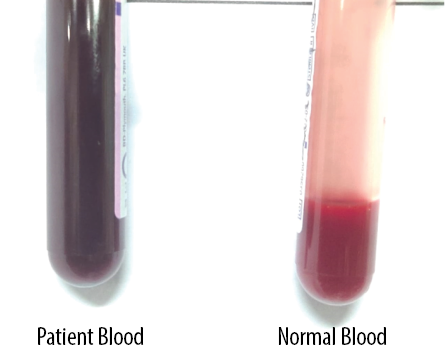 Figure 1: The patient’s blood sample (left) turned chocolate brown on exposure to 100% oxygen.
Figure 1: The patient’s blood sample (left) turned chocolate brown on exposure to 100% oxygen.
Discussion
The normal range of meth-Hb in blood is 1–3%. In methemoglobinemia, meth-Hb is generated in large amounts leading to decreased oxygen absorption in the lungs, which is exacerbated by the amount of generation of free radicals triggered by environmental conditions, dietary elements, or drug intake.
For congenital methemoglobinemia patients, usually no treatment is called for unless precipitated by drugs or environmental exposure. Acquired methemoglobinemia is much more dangerous and treatment is indicated due to the risk of impending neuro-cardiac symptoms. Congenital methemoglobinemia is so rare that its true incidence is unknown. It can occur due to autosomal recessive inheritance (leading to cytochrome b5 reductase deficiency) or dominant inheritance (leading to Hb M disease).5 Congenital methemoglobinemia can start in infancy and may present with a meth-Hb level of < 30% and compensatory erythrocytosis. They predominantly manifest as central cyanosis and other signs and symptoms can be aggravated by stress and oxidant exposure.4 Acute toxic methemoglobinemia: meth-Hb
> 20% triggers symptoms of hypoxia, and meth-Hb > 50% produces metabolic acidosis, coma, and
cardiac arrhythmia.
A blood sample turning chocolate brown on exposure to 100% oxygen is diagnostic of methemoglobinemia. In a low-resource setting, the quantitative color chart developed by Shihana et al,5 can be used to assess the concentration of meth-Hb in blood. Meth-Hb levels can be more accurately measured by advanced ABG, co-oximetry, spectrophotometry, or gas chromatography-mass spectrometry. Treatment is needed for acquired methemoglobinemia of > 30% for asymptomatic and > 20% for symptomatic patients. These cut-off levels can be lowered for anemic and
cardiac patients.
Congenital methemoglobinemia is a well-tolerated condition; treatment is needed in cases of oxidant stress. Methylene blue 5–7 mg/kg in 1–2 doses along with a high dose of ascorbic acid are given for symptomatic methemoglobinemia.3 Our patient’s positive family history in an immediate relative pointed towards congenital methemoglobinemia. Patients should avoid exposure to naphthalene-containing products, aniline dyes, and sulfa drugs. Oxidizing anesthesia drugs should be avoided during surgical treatment of such patients.
Positive response to ascorbic acid therapy in our patient suggested type 1 hereditary methemoglobinemia (autosomal recessive) with cytochrome b5 reductase deficiency.6,7 A confirmatory genetic testing could not be conducted due to a lack of facility in our institution and the patient’s financial constraints.
Conclusion
Dyshemoglobin disorders such as methemo-globinemia are rare blood conditions that can clinically manifest in a spectrum ranging from asymptomatic cyanosis to life-threatening cardiac dysfunction and can result in mortality if not recognized and intervened in time. Moreover, these disorders can be masked in congenital heart disease, and if unrecognized during surgery, the symptoms can get precipitated by oxidizing anesthetic agents.
Disclosure
The authors declared no conflicts of interests. Written informed consent was obtained from the patient’s parents for the publication of this report with images.
references
- 1. Todd JA, Aitman TJ, Cornall RJ, Ghosh S, Hall JR, 1. Rehman HU. Methemoglobinemia. West J Med 2001 Sep;175(3):193-196.
- 2. Lyle AN, Spurr R, Kirkey D, Albert CM, Billimoria Z, Perez J, et al. Case report of congenital methemoglobinemia: an uncommon cause of neonatal cyanosis. Matern Health Neonatol Perinatol 2022 Sep;8(1):7.
- 3. Beutler E, Lichtman MA, Coller BS, Kipps TJ. Methemoglobinemia and other causes of cyanosis. Williams Hematology 1994;5:654-662.
- 4. Aslan D, Türköz-Sucak G, Percy MJ. Recessive congenital methemoglobinemia in immediate generations. Turk J Pediatr 2016;58(1):113-115.
- 5. Shihana F, Dissanayake DM, Buckley NA, Dawson AH. A simple quantitative bedside test to determine methemoglobin. Annals of emergency medicine. 2010 Feb 1;55(2):184-9.
- 6. Guedri R, Missaoui N, Essaddam L, Ben Becher S. A rare cause of cyanosis: congenital methemoglobinemia. Clin Case Rep 2021 Jul;9(7):e04422.
- 7. Khade H, Raju U, Deshmukh T, Srivastav G, Kadiri V. Neonatal congenital methemoglobinemia–a case report. Indian J Med Case Rep 2016;5:12-17.