| |
Abstract
Proprotein convertase subtilisin/kexin type (PCSK9) is a crucial protein in LDL cholesterol (LDL-C) metabolism by virtue of its pivotal role in the degradation of the LDL receptor. Mutations in the PCSK9 gene have previously been found to segregate with autosomal dominant familial hypercholesterolemia (ADFH). In this study, DNA sequencing of the 12 exons of the PCSK9 gene has been performed for two patients with a clinical diagnosis of familial hypercholesterolemia where mutation in the LDL-receptor gene hasn't been excluded. One missense mutation was detected in the exon 9 PCSK9 gene in the two ADFH patients. The patients were found to be heterozygote for Ile474Val (SNP rs562556). Using an array of in silico tools, we have investigated the effect of the above mutation on different structural levels of the PCSK9 protein. Although, the mutation has already been reported in the literature for other populations, to the best of our knowledge this is the first report of a mutation in the PCSK9 gene from the Arab population, including the Omani population.
Keywords: Mutation; Autosomal dominant hypercholesterolemia; PCSK9 protien.
Introduction
Familial hypercholesterolemia (FH) is portrayed by elevated circulating concentrations of LDL particles which in turn increase the risk of cardiovascular disease.1 Mutations in the low density lipoprotein receptor (LDLR), apolipoprotein B (including Ag(x) antigen) (APOB), and proprotein convertase subtilisin/kexin 9 (PCSK9) genes are linked with FH1, FH2 and FH3, respectively.1,2 Two novel loci for hypercholesterolemia have been recognized on chromosome 16q22.1 (FH4) and 8q24.22; however, the cognate genes are yet to be identified.3,4
PCSK9, a secreted glycoprotein produced in the hepatocytes and enterocytes, interacts with the LDLR and mediates its lysosome-dependent degradation (Fig. 1).5 PCSK9 is synthesized in the endoplasmic reticulum (ER) as a preproprotein of 692 amino acid residues.6 As a pre-condition for its egress from the ER, PCSK9 auto-catalytically cleaves its pro-domain at VFAQ152¯SIP. The ~14-kDa pro-peptide and the ~62-kDa PCSK9 then form a heterodimer, which transits through the Golgi apparatus, is secreted, and interacts with the LDLR.7
The gene encoding PCSK9 is decidedly polymorphic. Two groups of PCSK9 sequence variants produce mild to unpretentious (and contrasting) phenotypes. Gain-of-function (GOF) sequence variants lead to a reduction in the LDLR that leads to hypercholesterolemia or to autosomal dominant hypercholesterolemia in cases of severe phenotypic variants.8 PCSK9 loss-of-function (LOF) sequence variants decrease LDLR degradation, thereby reducing LDL cholesterol (LDLC) concentrations.9 The important GOF and LOF mutations are shown in Fig. 1B.
A mutation in the PCSK9 gene has been identified across a number of populations of different ethnicities; however, its existence to the best of our knowledge is unknown in the Arab population, specifically in the Omani Arab population, although in recent times, a novel mutation in the LDLR gene hasn't been reported in an Omani family.10 In this study, DNA sequencing of the 12 exons of the PCSK9 gene has been performed for two patients with a clinical diagnosis of familial hypercholesterolemia where mutation in the LDL-receptor gene has been excluded. The patients were found to be heterozygous for I474V. The mutation is located in the C-terminal domain of the PCSK9 molecule (Fig. 1B) and has been previously reported, albeit not in the Omani Arab population. In order to obtain a comprehensive insight of the effect of these mutations on different structural levels of PCSK9, detailed bioinformatics analysis was carried out on the mutant protein.
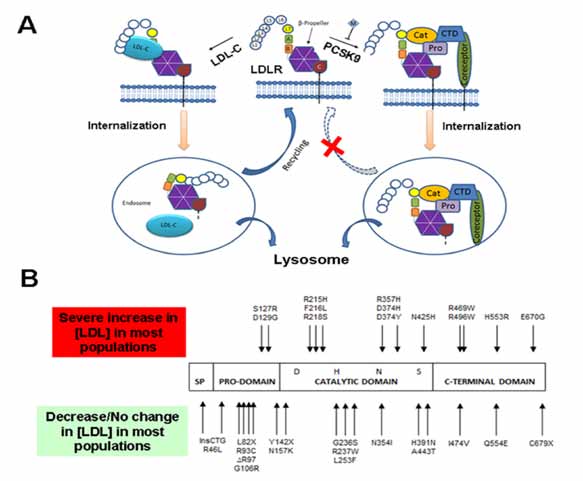
Figure 1: (A) Proposed mechanism for PCSK9-mediated LDLR down-regulation (Redrawn with minor modifications from Surdo et al., EMBO reports (2011) 12, 1300 - 1305: Top Center- LDLR on the cell surface at pH 7.0 (neutral pH); Left- Under normal conditions, LDL-cholesterol binds to the LDLR and the whole complex is internalized in an endosome. LDL-cholesterol is degraded in lysosomes and the LDLR is recycled and returned to the surface for again binding to the LDL-cholesterol and thus helping in cholesterol homeostasis. Right- When PCSK9 binds to the LDLR, the whole complex is internalized in an endosome. The endosome further fuses with the lysosome and the PCSK9-LDLR complex is degraded. This degradation of LDLR prevents recycling of LDLR which in turn disrupts cholesterol homeostasis and leads to Familial Hypercholesterolemia. CTD: C-Terminal histidine-rich Domain; EGFPH: Epidermal Growth Factor Precursor Homology domain; LBD: Ligand-Binding Domain; LDL-c: Low-Density Lipoprotein-Cholesterol; LDLR, Low-Density Lipoprotein Receptor; PCSK9: Proprotein Convertase Subtilisin/Kexin type 9. (B) The PCSK9 domain architecture showing the four domains: Signaling Peptide (SP), Pro-domain, catalytic domain and C-terminal domain. The mutations at the top are GOF mutations and thus increase LDL (lead to hypercholesterolemia) whereas the mutations at the bottom are LOF mutations which lead to the reduction of LDL levels in the body (lead to hypocholesterolemia).
Case Reports
The two patients presented in this study were diagnosed with FH based on the Simon Broome criteria. Patient one (male) pre-treatment lipid profile indicated the following: total cholesterol 18.2 mmol/l; low density lipoprotein cholesterol (LDL-C) 16.6 mmol/l; triglyceride 0.68 mmol/l; apolipoprotein B (ApoB) 4.4 g/l. He had no history of coronary artery disease but was diagnosed with diabetes mellitus and was on insulin injection, rousvastatin 40 mg, ezetimibe 10 mg and biweekly LDL-apheresis performed using a DALI (Direct Adsorption of Lipoproteins) system (Fresenius SE & Co. KGaA). The patient responded well to the combination of lipid lowering therapy and the LDL-apheresis with an average LDL-C reduction of 62% post-therapy.
Patient two (female) was the sister of patient one, her lipid profile pre-treatment indicated the following: total cholesterol 17.8 mmol/l; low density lipoprotein cholesterol (LDL-C) 15.2 mmol/l; triglyceride 1.8 mmol/l; apolipoprotein B (ApoB) 3.8 g/l. She also had history of severe carotid atherosclerosis and underwent right endarterectomy (surgical technique to eradicate the atheromatous plaque material). She was treated with combination of rousvastatin 40 mg, ezetimibe 10 mg and biweekly LDL-Apheresis performed using a DALI-system. The average LDL-C reduction was 60% post-therapy.
In terms of DNA sequencing of individual exons of the PCSK9 gene; both patients did not possess a mutation in the LDL receptor gene that could possibly affect the function of the LDLR, as determined by DNA sequencing of the translated parts of the 18 exons of LDLR gene. Primer sequences for the amplification of the 12 exons of the PCSK9 gene are summarized in the study by Abifadel et al.2 Standard DNA-sequencing reactions using version 3.1 of Big Dye Terminator cycle sequencing kit (Applied Biosystems, Foster City, CA) were analyzed on a Genetic Analyzer 3100 (Applied Biosystems, Foster City, CA). Nucleotide positions of cDNA were numbered according to the published sequence (accession number NM 174936) with A of the ATG translation initiation codon being nucleotide 1. The obtained gene sequence was translated to protein using the Translate software module available at http://us.expasy.org/tools/#translate
For the analysis of Protein Structure; hydropathy analysis using the Kyte-Doolittle algorithm,11 was performed using a window size of nine amino acids using linear weight variation model, for both mutant and wild type PCSK9. The three-dimensional structure of I474V-PCSK9 was modeled using the standard alignment routine of SWISS-MODEL program.12 The known crystal structure of the wild type PCSK9 in complex with the EGF-A domain of LDLR (PDB identifier 3GCX) was used to construct the homology-based models.13 The template structure was selected on the basis of highest sequence similarity. Validation was performed by evaluating the stereochemical feasibility of the torsion angles using the Ramachandran Plot (RP),14 generated using the graphic package Ramachandran Plot (version 2) hosted on Indian Institute of Science, Bangalore server.
Identification of Mutation in the PCSK9 gene was done by analyzing the obtained gene sequences for both patients one (male) and two (female) which indicated the presence of a heterozygous non-synonymous missense mutation in exon 9 (Fig. 2A- representative raw sequence data), that results in the substitution of isoleucine with valine at position 474 in the primary structure of the protein.
The effect of the mutation on protein hydropathy was investigated using Kyte-Doolittle algorithm. In order to generate data for such an analysis, the entire protein sequences of both wild type and I474V-PCSK9 were scanned with a moving window of a definitive size (9 amino acids). At each position, the mean hydrophobic index of the amino acids within the window was calculated and that value plotted as the midpoint of the window. The aim was to investigate, if the presence of the mutation affects hydrophobicity of the neighboring amino acids including the site of mutation. A value >0 indicates a hydrophobic region. As evident in Fig. 2B, presence of the mutation affects the hydrophobicity at amino acid positions 470, 471, 472, 473, 475, 476, 477 and 478 including the point of mutation at 474. In I474V PCSK9, there is a decrease in hydrophobicity at 470, 471, 472, 473, 474 and 475. At position 476, there is a reversal in the hydropathy, from hydrophobic and hydrophilic for the I474V PCSK9 as the score here <0 versus >0 in the wild-type PCSK9. Following this reversal, there is a concomitant increase in hydrophilicity at positions 476 and 477 for I474V PCSK9.
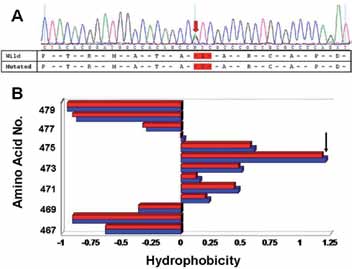
Figure 2: (A) DNA analysis: Representative sequence data highlighting the presence of a missense mutation in the exon 9 of ADFH patient one. (B) Kyte-Doolittle hydropathy analysis of amino acids for wild type (Blue) and I474V-PCSK9 (red) surrounding the point of mutation (the exact point of mutation is indicated by an arrow). Note: At position 476, there is a reversal in the hydropathy, from hydrophobic to hydrophilic for the I474V-PCSK9 as the score here is <0 versus >0 in the wild-type PCSK9.
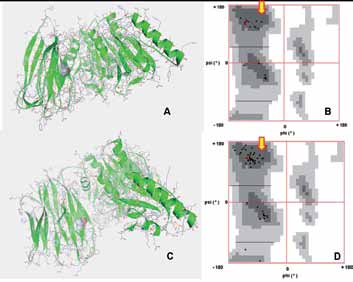
Figure 3: (A) Structure of the wild type PCSK9. The structural coordinates were obtained from the PDB (ID: 3GCX) and plotted in Python graphics package. The presence of Ile at 474 is shown (CPK representation) with respect to other amino acids (stick representation) in PCSK9. (B) Ramachandran plot (RP) showing the presence of the Ile474 (red dot) in a predominantly beta-sheeted region. (C) Homology modeled structure of Ile474Val-PCSK9. The presence of Val at 474 is shown (CPK representation) with respect to other amino acids (stick representation) in PCSK9. Note: The overall structure of PCSK9 in the mutant is similar to the wild type protein. (D) RP plot showing the presence of Val474 (red dot) in a predominantly beta-sheeted region of the protein. Note: The presence of the mutant residue Val in the beta-sheeted region similar to the wild type Ile reflects that there is not much alteration in the local folding around the site of mutation.
In terms of the effect of the mutation on the tertiary structure of PCSK9; the modeled structure of I474V-PCSK9 (Fig. 3C) did not exhibit any drastic alteration in the tertiary structure with respect to the wild type PCSK9 (Fig. 3A). In order to predict the validity of the model, RP analysis was performed. This plot displays backbone dihedral angles ψ against φ of amino acid residues in protein structure. For the modeled structure of I474 PCSK9 94.8% of the total amino acid residues are in the most favored region of the RP with an additional 5.0% in the allowed regions (Plot not shown). This reflects that the obtained model of I474V-PCSK9 has a high reliability. Further, we checked the effect of the mutation on the local conformation PCSK9 using RP. The upper left quadrant of the RP (indicated by the block arrows in Fig. 3B and D) corresponds to beta-sheet. Therefore, in the wild type protein the isoleucine at 474th position (indicated by the red dot in Fig. 3B) is localized in the region of PCSK9 which predominantly exhibits a beta-sheeted fold. In I474V-PCSK9, the mutated valine at the 474th position (indicated by the red dot in Fig. 3D) is also situated in a predominantly beta-sheeted region of the protein. This indicates that the mutation of isoleucine to valine at 474, plausibly does not affect the local conformation drastically.
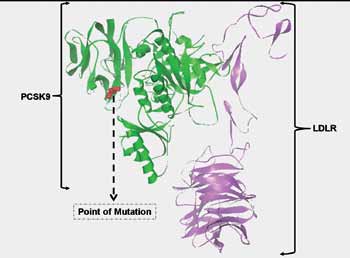
Figure 4: The site of mutation with respect to the PCSK9-LDLR complex tertiary structure. The structural coordinates were obtained from PDB (ID: 3P5C) and plotted in Python graphics package. Note: The site of mutation is away from the site of interaction of PCSK9 with LDLR.
Discussion
This study reports the presence of a missense mutation in the PCSK9 gene in two Omani Arab patients diagnosed with ADFH, where mutation in the LDLR gene hasn't been excluded. The exon 9 mutation leads to the substitution of isoleucine at 474th position to valine (Fig. 2A), and is located in the C-terminal domain of the protein (Fig. 1B). To the best of our knowledge this is the first report of a mutation in the PCSK9 gene from the Arab population, including the Omani population. The I474V-PCSK9 variant has been found in several other populations. Interestingly, I474V in most populations is not associated with any changes in LDLC concentration.15,16 However in the general population in Japan the exon9/I474V mutation along with intron 1/C(-161)T polymorphism were shown to be significantly associated with increased total cholesterol and LDLC levels.17,18 We are in the process of confirming if a similar intronic polymorphism as observed in the Japanese population is also present in the Omani Arab patients with the PCSK9-I474V variant.
In a recent study, investigating evidence for positive selection in the C-terminal domain of PCSK9 gene based on phylogenetic analysis across 14 primate species, a striking pattern of I474V variation across the primates was observed.19 The ancestral state of the 474th amino acid (M or V) in New World monkeys is not clearly delineated, as an out-group is non-existent. Further, as depicted in the article, the ‘V’ allele diverged to ‘I’ or ‘V’ in the Hominoid clade, suggesting a dynamic evolutionary history of the 474th amino acid. The human mutation I to V replicates the ancestral state, and the recurrence of this ancestral state has functional consequences across different populations. In summary, I474V variant of PCSK9 may detrimentally affect the total cholesterol and LCLC levels in association with other compounding mutations. We have already eliminated that these patients do not have any mutation in the LDLR gene and need to rule out mutations APOB gene, which we are also in the process of checking. However, in the absence of mutations in the known genes associated with ADFH, possibility cannot be ruled out that this mutation compounded with a mutation present in another loc(i)us (for which the cognate gene(s) remain undiscovered) adversely affect the total cholesterol and LDLC. Exome sequencing strategy in line with that recently availed by Hussin et al. may provide a concrete answer to the above.20
We investigated the effect of the mutation on the hydropathy of PCSK9. At position 476, there is a reversal in the hydropathy, from hydrophobic to hydrophilic for the I474V-PCSK9 as the score here is <0 versus >0 in the wild-type PCSK9. Following this reversal, there is a concomitant increase in hydrophilicity at positions 476 and 477 for I474V-PCSK9 (Fig. 2B). However this change is not as drastic as those observed for some of the GOF-mutations such as D374Y-PCSK9 (Shah K and Banerjee Y, manuscript under preparation). In order to see if the mutation will affect the tertiary structure of PCSK9, we homology modeled the mutant. The presence of the mutation does not alter the overall tertiary structure of the protein with respect to the wild type (Figs. 3A and C). Further local folding of PCSK9 in and around the site of mutation remains unperturbed, as is evident from RP analysis (Figs. 3B and D). Additionally, the site of mutation is localized away from the site of LDLR binding (Fig. 4), which indicates towards the possibility that the mutation may not increase or decrease the binding affinity of PCSK9 towards LDLR. However, real-time binding experiments involving surface plasmon resonance and isothermal titration calorimetry, will provide a tangible answer towards this effect.
Conclusion
In summary, this is the first report of a mutation in the PCSK9 gene discovered in Omani Arab ADFH subjects. However, further studies with regards to the (A) elucidation of the segregation of the mutation in the Omani population; and (B) compounding effect of the mutation, with known mutations in PCSK9 intron and APOB gene (if present), need to be investigated. Lastly, structure-activity relationship studies of I474V-PCSK9 versus the wild-type PCSK9 will facilitate the augmentation of knowledge with regards to PCSK9’s role in cholesterol metabolism.
Acknowledgements
YB and KR are recipient of SQU college grant IG/MED/BIOC/12/01 which supported this study. WZ is a recipient of SQU MSc fellowship award. YB thanks Dr. Peter Lansberg, Durrer Cardiovascular Genetic Research Center, Netherlands for helpful discussions. Authors declare no competing interest.
References
1. Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol 2004 Sep;160(5):407-420.
2. Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003 Jun;34(2):154-156.
3. Marques-Pinheiro A, Marduel M, Rabès JP, Devillers M, Villéger L, Allard D, et al; French Research Network on ADH. A fourth locus for autosomal dominant hypercholesterolemia maps at 16q22.1. Eur J Hum Genet 2010 Nov;18(11):1236-1242.
4. Cenarro A, García-Otín AL, Tejedor MT, Solanas M, Jarauta E, Junquera C, et al. A presumptive new locus for autosomal dominant hypercholesterolemia mapping to 8q24.22. Clin Genet 2011 May;79(5):475-481.
5. Banerjee Y, Shah K, Al-Rasadi K. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med 2012 Jun;366(25):2425-2426, author reply 2426.
6. Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A 2003 Feb;100(3):928-933.
7. Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem 2004 Nov;279(47):48865-48875.
8. Allard D, Amsellem S, Abifadel M, Trillard M, Devillers M, Luc G, et al. Novel mutations of the PCSK9 gene cause variable phenotype of autosomal dominant hypercholesterolemia. Hum Mutat 2005 Nov;26(5):497.
9. Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet 2005 Feb;37(2):161-165.
10. Al-Hinai AT, Al-Abri A, Al-Dhuhli H, Al-Waili K, Al-Sabti H, Al-Yaarubi S, et al. First Case Report of Familial Hypercholesterolemia in an Omani Family due to Novel Mutation in the Low-Density Lipoprotein Receptor Gene. Angiology 2012 Nov.
11. Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol 1982 May;157(1):105-132.
12. Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res 2009 Jan;37(Database issue):D387-D392.
13. McNutt MC, Kwon HJ, Chen C, Chen JR, Horton JD, Lagace TA. Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J Biol Chem 2009 Apr;284(16):10561-10570.
14. Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol 1963 Jul;7:95-99.
15. Kotowski IK, Pertsemlidis A, Luke A, Cooper RS, Vega GL, Cohen JC, et al. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet 2006 Mar;78(3):410-422.
16. Miyake Y, Kimura R, Kokubo Y, Okayama A, Tomoike H, Yamamura T, et al. Genetic variants in PCSK9 in the Japanese population: rare genetic variants in PCSK9 might collectively contribute to plasma LDL cholesterol levels in the general population. Atherosclerosis 2008 Jan;196(1):29-36.
17. Shioji K, Mannami T, Kokubo Y, Inamoto N, Takagi S, Goto Y, et al. Genetic variants in PCSK9 affect the cholesterol level in Japanese. J Hum Genet 2004;49(2):109-114.
18. Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem 2004 Nov;279(47):48865-48875.
19. Ding K, McDonough SJ, Kullo IJ. Evidence for positive selection in the C-terminal domain of the cholesterol metabolism gene PCSK9 based on phylogenetic analysis in 14 primate species. PLoS One 2007;2(10):e1098.
20. Hussin J, Sinnett D, Casals F, Idaghdour Y, Bruat V, Saillour V, et al. Rare allelic forms of PRDM9 associated with childhood leukemogenesis. Genome Res 2012 Dec.
|
|