Neurofibromatosis type 1 (NF-1), also known as von Recklinghausen disease, results from a mutation of the NF-1 gene inherited via an autosomal dominant Mendelian pattern. NF-1 is the most common subtype of neurofibromatosis typically diagnosed in the first decade of life.1 NF-1 is characterized by developing multiple non-cancerous nerve and skin tumors (neurofibromas), Lisch nodules, café-au-lait, and axillary and inguinal freckling.2,3
The presence of the NF-1 mutation gives a 34-times increased risk of developing sarcoma compared to the general population.4 Neurosarcoma is another common malignant transformation among NF-1.4
Spindle cell sarcoma is a malignant soft tissue tumor identified by the presence of spindle shape neoplastic cells. It could be locally infiltrative with metastatic potential, and the recurrences commonly occurs within 24 months of the index resection.5 Spindle cell sarcoma is subtyped via genetics, immunophenotype, morphology, and degree of differentiation, and broadly divided into differentiated types (leiomyosarcoma, fibrosarcoma, and myofibroblastic sarcoma) and uncertain differentiation type (synovial sarcoma).6 The incidence of spindle cell sarcoma in the perineum with a background of NF-1 is rare, with < 1% of all soft tissue sarcoma.5 We report a case of solitary spindle cell sarcoma presenting at the perineum of a lady with NF-1.
Case report
A 47-year-old female with a background of NF-1 presented to our surgical department with an eight-month history of rapidly increasing left perineal swelling and pain upon sitting. The swelling was initially small (2 × 1 cm); however, it rapidly grew to 10 × 12 cm over eight months. In addition, her mother and younger brother were also diagnosed with NF-1.
On examination, there was a mass located from one to six o’clock position, 4 cm away from the anal verge [Figure 1].
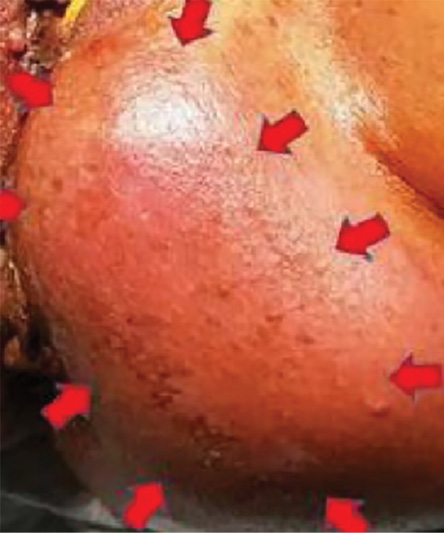 Figure 1: Perineal mass measuring 10 × 12 cm from one to six o’clock position around the perineum (outlined by red arrows).
Figure 1: Perineal mass measuring 10 × 12 cm from one to six o’clock position around the perineum (outlined by red arrows).
Colonoscopy was normal until cecum with no intraluminal mucosal lesion seen. The rapidly growing mass was highly suspicious of malignancy. Contrast-enhanced computed tomography of the thorax, abdomen, and pelvis was performed and confirmed no distant metastases. Magnetic resonance imaging (MRI) of the pelvis reported a soft tissue mass measuring approximately 5.2 × 10.2 × 5.2 cm (anteroposterior × width × craniocaudal), displacing the vagina and anus to the right with no direct local infiltration to the adjacent organs. It appeared to be a tumor of mesenchymal origin with the differentials of liposarcoma [Figure 2].
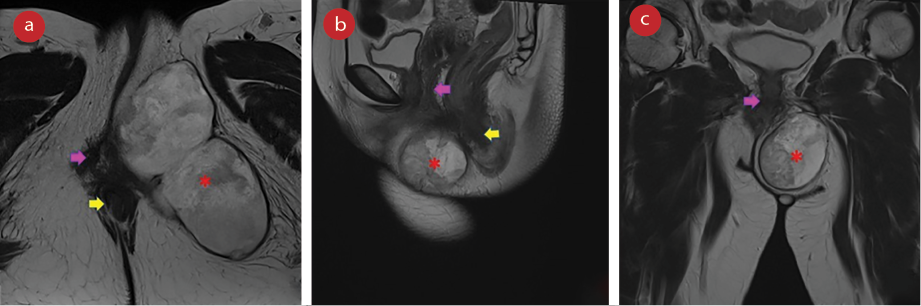 Figure 2: MRI of the pelvis T2-weighted images in (a) axial, (b) sagittal, and (c) coronal view demonstrating the perineal mass (red asterisks) compressing and displacing the vagina (pink arrows), and anus (yellow arrows).
Figure 2: MRI of the pelvis T2-weighted images in (a) axial, (b) sagittal, and (c) coronal view demonstrating the perineal mass (red asterisks) compressing and displacing the vagina (pink arrows), and anus (yellow arrows).
Ultrasound-guided core-needle biopsy confirmed a spindle cell lesion. She underwent wide local excision of the left perineal mass. The intraoperative finding was a 10 cm × 12 cm irregular border soft-tissue mass at the left perineal area with attachment to the overlying skin but no infiltration to adjacent bone and external anal sphincter [Figure 3]. The elliptical skin incision was closed without tension and did not require further reconstruction. She was discharged one day postoperatively.
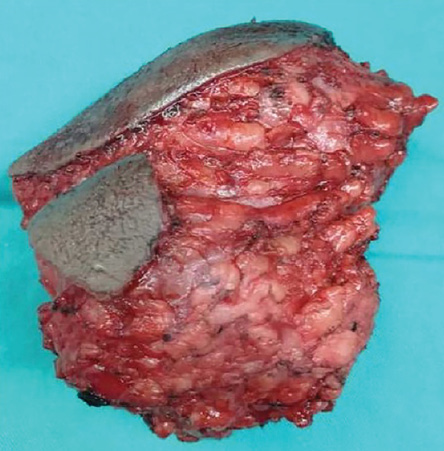 Figure 3: A 10 × 12 cm irregular shaped tumor removed from the left perineum with attachment to the skin, but no local infiltration to bone and
Figure 3: A 10 × 12 cm irregular shaped tumor removed from the left perineum with attachment to the skin, but no local infiltration to bone and
anal sphincter.
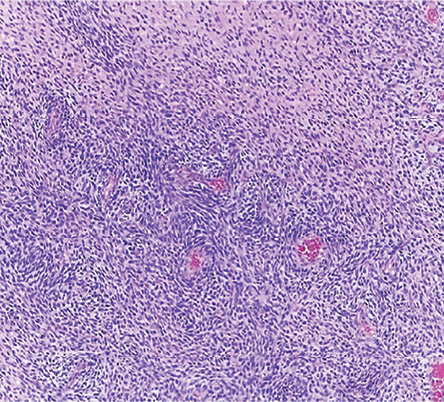 Figure 4: Fascicles of spindle-shaped cells with varying cellularity and perivascular accentuation (hematoxylin and eosin stain, magnification
Figure 4: Fascicles of spindle-shaped cells with varying cellularity and perivascular accentuation (hematoxylin and eosin stain, magnification
= 100 ×).
The histopathological examination revealed a malignant spindle cell tumor with a clear 30 mm surgical margin and high-grade sarcoma [Figure 4]. At one month follow-up, her left perineal scar was well-healed. She was also referred to the radiation-oncology team for radiotherapy. She visited at three and six-months follow-up with no evidence of local recurrence on surveillance contrast-enhanced computed tomography of the thorax, abdomen, and pelvis.
Discussion
NF-1 is at higher risk of developing malignant soft tissue tumors, and this incidence rises with advancing age; 2% for an individual under 21 and 4.2% for individuals over 21.7 Soft tissue sarcomas such as angiosarcoma, chondrosarcoma, rhabdosarcoma, leiomyosarcoma, and undifferentiated pleomorphic sarcoma were more common among NF-1 patients in their sixth and seventh decades of life; malignant peripheral nerve tumors were a more common occurrence in their younger counterparts.8
The predilection of tumor development to either the benign or malignant variant in NF-1 imposed a significant diagnostic challenge. This is crucial to prognostication and treatment planning. Moreover, it gets more challenging to diagnose when the soft tissue tumor presents in an unusual area such as the perineum—nevertheless, there are reports of these tumors presenting in the female genitalia and genitourinary system.9,10
Soft tissue sarcoma of spindle cell variant was classified into two types; clear differentiation lineage type such as leiomyosarcoma and fibrosarcoma, and the uncertain lineage type.11,12 Perineum spindle cell sarcoma was rare, and due to its heterogenicity, it imposed a significant diagnostic challenge.
A multidisciplinary approach to spindle cell sarcoma remains a challenging endeavor despite the advances in cytogenetics and immunohistochemistry. MRI remains the imaging of choice for soft tissue tumors and is essential for surgical planning and staging. Biopsy has a limited role as most of these tumors eventually require resection. The biopsy is reserved for the tumor that is potentially responding to preoperative radiotherapy or chemotherapy.
Wide local surgical excision with clear margin remains the gold-standard treatment for soft tissue sarcoma. The close or unclear margins have a proportional relationship to the recurrence rate and poorer prognosis.13 The soft tissue tumor management strategy revolutionized as we researched and explored in-depth its molecular biology and immuno-profiling. Most of these soft tissue sarcomas were angiogenesis-dependent tumors. Anti-angiogenesis therapy such as pazopanib, sorafenib, and sunitinib was recommended in the latest National Comprehensive Cancer Network guidelines as targeted therapy for advanced or metastatic soft tissue sarcoma.1,7,13 On going studies exploring the role of immunotherapy in soft tissue sarcoma produced promising results but were heterogeneous. The immune profiling and identification of immune biomarkers like the immune checkpoint inhibitors in soft tissue sarcoma were challenging due to its rare and heterogeneous disease.14
Tissue biopsy has a limited role in the management of soft tissue tumors. However, in NF-1 patients, attaining information regarding benign or malignant nature facilitates planning to the extent of resection. Molecular testing on angiogenesis-dependent factors may help us decide which patients should be subjected to adjuvant immunotherapy selectively. Adjuvant radiotherapy is currently the most advocated preventive strategy toward recurrence.
Conclusion
A malignant spindle cell tumor at the perineum is a rare variant of high-grade sarcoma. Soft tissue neoplasms in NF-1 present a diagnostic challenge in discriminating those with malignant potential. Therefore, wide local excision complemented with adjuvant radiation therapy is the best strategy to reduce recurrence. In addition, there was emerging promising evidence from adjuvant chemotherapy and targeted therapy depending on the variant of the soft tissue sarcoma.
Disclosure
The authors declared no conflicts of interest. A written consent was obtained for the publication of this case report.
references
- 1. Zhang Y, Chao J-J, Liu X-F, Qin S-K. Type I neurofibromatosis with spindle cell sarcoma: a case report. World J Clin Cases 2019 Oct;7(19):3104-3110.
- 2. Brosius S. A history of von Recklinghausen’s NF1. J Hist Neurosci 2010 Oct;19(4):333-348.
- 3. Brems H, Beert E, de Ravel T, Legius E. Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol 2009 May;10(5):508-515.
- 4. Yap YS, McPherson JR, Ong CK, Rozen SG, Teh BT, Lee AS, et al. The NF1 gene revisited - from bench to bedside. Oncotarget 2014 Aug;5(15):5873-5892.
- 5. Levy AD, Manning MA, Miettinen MM. Soft-tissue sarcomas of the abdomen and pelvis: Radiologic-pathologic features, part 2— uncommon sarcomas. Radiographics 2017 May-Jun;37(3):797-812.
- 6. Collini P, Sorensen PH, Patel S, Blay JY, Issels RD, Maki RG, et al. Sarcomas with spindle cell morphology. Semin Oncol 2009 Aug;36(4):324-337.
- 7. Von Mehren M, Randall RL, Benjamin RS, Boles S, Bui MM, Conrad EU III, et al. Soft tissue sarcoma, version 2.2016 clinical practice guidelines in oncology NCCN categories of evidence and consensus. JNCCN-Journal Natl Compr Cancer Netw 2016;14(6):758-786.
- 8. Stojadinovic A, Leung DH, Hoos A, Jaques DP, Lewis JJ, Brennan MF. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tissue sarcomas. Ann Surg 2002 Mar;235(3):424-434.
- 9. Musella A, Palaia I, Domenici L, Casorelli A, Martoccia A, Benedetti Panici P. Vulvar malignancy in neurofibromatosis syndrome. Case Rep Obstet Gynecol 2013;2013:217924.
- 10. Saquib S, Cherawala M, Abdel Rahman O, Keloth TE. Leiomyosarcoma of the vulva mimicking as chronic bartholin cyst: a case report. Oman Med J 2020 Jul;35(4):e153.
- 11. Nystrom LM, Reimer NB, Reith JD, Dang L, Zlotecki RA, Scarborough MT, et al. Multidisciplinary management of soft tissue sarcoma. ScientificWorldJournal 2013 Jul;2013:852462.
- 12. Ang CY, Kau CY, Chew GK, Tan MH. A misdiagnosed spindle cell sarcoma. Proc Singapore Healthc. 2013;22(4):273-276.
- 13. DuBois S, Demetri G. Markers of angiogenesis and clinical features in patients with sarcoma. Cancer 2007 Mar;109(5):813-819.
- 14. Clemente O, Ottaiano A, Di Lorenzo G, Bracigliano A, Lamia S, Cannella L, et al. Is immunotherapy in the future of therapeutic management of sarcomas? J Transl Med 2021 Apr;19(1):173.