Pulmonary alveolar microlithiasis (PAM) is an uncommon disorder characterized by calcium phosphatase crystals deposition into the alveoli.1 PAM was described for the first time by Marcello Malpighi in 1868, and the histopathology was detailed by Harbitz in 1918.2 PAM is caused by a mutation in the SLC34A2 gene encoding the type IIb sodium phosphate cotransporter in alveolar type II cells.3 Early in the disease, most patients are asymptomatic, and the disease might only be detected incidentally on chest radiograph performed for other reasons. As the disease progresses, some patients develop symptoms such as cough and shortness of breath and can progress to pulmonary fibrosis, respiratory failure, and cor pulmonale.1–3 The typical findings on chest radiographs are bilateral fine sand-like micronodules of calcific density with a sandstorm appearance that involves mainly the middle and lower lung zones.4
Here, we report the case of a 33-year-old Omani male with PAM with classical radiological findings. To the best of our knowledge, this the first case of PAM to be reported in the Omani population.
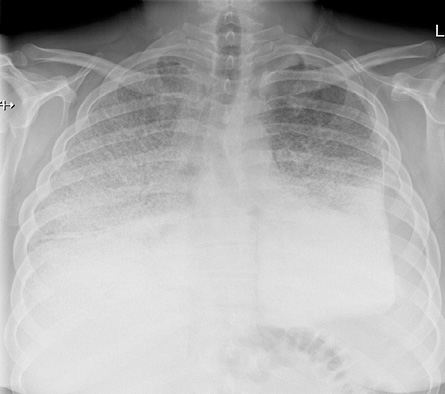
Figure 1: Frontal chest radiograph showing bilateral mid to upper lung zone dense micronodular patter with reticulation along with bilateral lower lung zone dense consolidation. There is a small left sided pleural effusion.
Case report
A 33-year-old Omani male presented to a local health center for the first time in July 2017 with a one-week history of fever and shortness of breath. The patient was then referred to our institute, Royal Hospital, Muscat, for further workup. Upon arrival, the patient was afebrile (36.4 oC). However, he was sick looking and tachypneic. His respiratory rate was 44 with a high blood pressure (150/100 mmHg). His saturation was 75% in room air. On auscultation, he had bilateral crepitation. Laparotomy examinations showed high C-reactive protein (52 mg/L). Arterial blood gas analysis showed: pH 7.33, carbon dioxide partial pressure 62 mmHg, oxygen partial pressure 83 mmHg, and fraction of inspired oxygen 100. His complete blood count was normal, and his white blood cell count was 9.4 × 109/L (normal range: 7.2–10.5 × 109/L), blood platelet count was 243 × 109/L (normal range: 140–400 × 109/L), hemoglobin levels were 14.4 g/dL (normal range: 11.5–15.5 g/dL), calcium was 2.33 mmol/L (normal range: 2.1–2.55 mmol/L), corrected calcium for albumin was 2.59 mmol/L (normal range: 2.1–2.6 mmol/L), phosphate was 0.84 mol/L (normal range: 0.75–1.5 mol/L), and parathyroid was 2.9 pmol/L (normal range: 1.6–6.9 pmol/L). Other lab investigations were within normal limits including renal and liver function tests. A chest radiograph was performed and revealed bilateral dense consolidation [Figure 1].
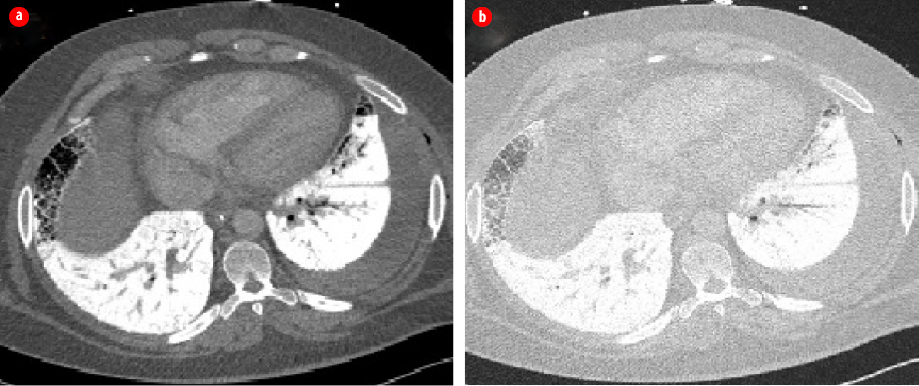
Figure 2: High-resolution chest computed tomography scans (a) axial mediastinal window and
(b) lung window showing bilateral dense interlobular thickening and extensive consolidations
with calcific density.
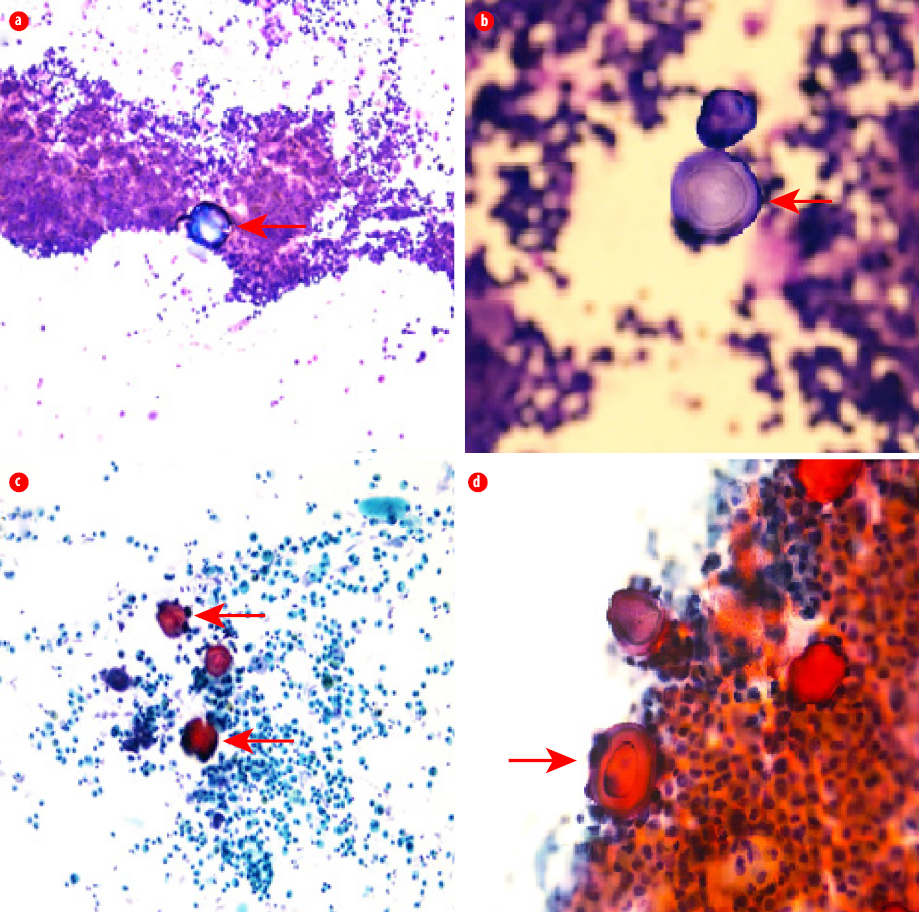
Figure 3: Bronchoalveolar lavage. (a) May-Grünwald-Giemsa stain, magnification = 40 × (red arrow).
(b) A lamellar concentric microlith (red arrow) in bronchoalveolar lavage, magnification = 100 ×.
(c) Papanicolaou stain, magnification = 40 ×. (d) Multiple concentric microliths in bronchoalveolar lavage, magnification = 100 × (red arrow).
The impression was severe lower respiratory tract infection complicated by acute respiratory distress syndrome (ARDS). The patient was electively intubated and admitted to the intensive care unit. Septic workup including respiratory viral panel, human immunodeficiency virus, sputum, urine, and blood cultures were negative. Sputum was negative for acid-fast bacilli. Further evaluation of chest radiograph findings with high-resolution computed tomography (HRCT) of the chest showed bilateral dense interlobular thickening and extensive consolidations with a calcific density distributed bilaterally throughout both lung fields with a lower lobe predominance [Figure 2], findings were highly suggestive of PAM. To confirm the diagnosis, bronchoscopy and bronchoalveolar lavage were performed. Mycobacterium tuberculosis culture was negative, and no other pathogens were isolated. Histopathological examination showed cellular smears composed of scattered lamellar microliths accompanied by many foamy macrophages and some bronchial epithelial cells [Figure 3]. The findings were consistent with the diagnosis of PAM. The patient continued to be treated as a lower respiratory tract infection and made a full recovery. However, he continues to complain of shortness of breath on exertion. The treatment for PAM is lung transplant. However, lung transplant was not feasible in our patient, and we chose to treat him symptomatically. The patient had genetic counseling, but no genetic testing was done as the test was not available.
Discussion
PAM is a rare heredity disease of the lung that is characterized by a chronic intra-alveolar accumulation of microliths.6 It is caused by a mutation in the SLC34A2 gene encoding type IIb sodium phosphate cotransporter in alveolar type II cells.7 This leads to reduced uptake of phosphate, which causes the formation of intra-alveolar microliths as a result of phosphate chelating calcium in extracellular fluid.2 Although PAM can affect all age groups, most of the affected patients are in their second and third decades of life. PAM has a slight male predominance, and only 37% of patients have a positive family history of PAM.3 Our patient was an Omani male in his third decade of life with no family history of PAM.
Early in the disease, patients with PAM are either asymptomatic or might experience mild symptoms such as non-productive cough or shortness of breath. After the diagnosis, the course of the disease varies, while in a few patients the radiological findings and symptoms remain stable. The majority of patients have a slowly progressive course that eventually ends into pulmonary fibrosis, cor pulmonale, and respiratory failure.1,2,4,6,8
The diagnosis of PAM is usually established by identifying the typical radiological and histopathological features. Radiological findings depend on the severity of the disease. On chest radiographs, patients with PAM usually have bilateral, sand-like micronodules of calcific density with a sandstorm appearance predominantly in the mid and lower lung zones.2,3 On HRCT, more subtle findings can be detected, which might not be best appreciated on the chest radiograph. Early in the disease, bilateral non-calcified micronodules can be seen in the HRCT of asymptomatic children with PAM. As the disease progresses, there are usually bilateral innumerable calcified micronodules (< 1 mm) distributed throughout both lung fields with predominant involvement of the mid to lower lung zones and posterior basal segments.3,4,8,9 These calcified micronodules increase in size and density and coalescent together to form calcified air space consolidation and ground-glass opacities. This is usually associated with calcified micronodular interlobular septal thickening giving a ‘crazy paving’ appearance.8 Crazy paving can be seen in many other conditions, including pulmonary infection, pulmonary hemorrhage, ARDS, and alveolar proteinases. In PAM, interlobular septal thickening is usually of calcific density. Other typical findings are apical bullae and bilateral subpleural cystic changes that cause what is known as black pleura sign, which can be seen on chest radiograph and HRCT.2
Although the diagnosis of PAM can be established radiologically, bronchoalveolar lavage or lung biopsy might be required to confirm the diagnosis by demonstrating microliths that are typically periodic acid-Schiff positive and composed of centric calcareous lamellae around a central nucleus.3,4,8–10 Our patient had a bronchoalveolar lavage, which showed cellular smears composed of scattered lamellar microliths accompanied by many foamy macrophages and some bronchial epithelial cells. Genotyping for SLC34A2 gene mutations can be useful for screening family members, with appropriate genetic counseling. However, it is not currently clinically informative or necessary for diagnosis.1,3,4,8
Since the SLC34A2 gene is expressed by extrapulmonary tissue such as kidneys, prostate, testicles, seminal vesicles, pancreas, and liver, patients with PAM can have extrapulmonary calcification such as medullary nephrocalcinosis, nephrolithiasis, and calcification of the seminal vesicles.3,4,7 Azoospermia due to epididymal and periurethral calcification has been reported.11 Our patient did not have extrapulmonary findings.
Currently, there is no effective treatment for PAM. Corticosteroid, calcium chelating agent, and bronchoalveolar lavage have been used to treat and to slow the progression, and none are reported to be effective. Lung transplant remains the only effective treatment for PAM with no reported recurrence following transplantation.3,8,10
Conclusion
PAM is uncommon heredity disorder, which needs a high index of suspicion. Awareness of typical radiological and pathological findings of PAM will help clinicians, radiologists, and pathologists to make the correct diagnosis.
Disclosure
The authors declared no conflicts of interest.
references
- 1. Zhang X-D, Gao JM, Luo JM, Zhao Y. Pulmonary alveolar microlithiasis: a case report and review of the literature. Exp Ther Med 2018 Jan;15(1):831-837.
- 2. Khaladkar SM, Kondapavuluri SK, Kamal A, Kalra R, Kuber R. Pulmonary alveolar microlithiasis - clinico-radiological dissociation - a case report with radiological review. J Radiol Case Rep 2016 Jan;10(1):14-21.
- 3. Castellana G, Castellana G, Gentile M, Castellana R, Resta O. Pulmonary alveolar microlithiasis: review of the 1022 cases reported worldwide. Eur Respir Rev 2015 Dec;24(138):607-620.
- 4. Khurana A, Malik R, Sharma J, Khurana U, Joshi D, Goyal A. Pulmonary alveolar microlithiasis: a commonly misdiagnosed rare entity. Sultan Qaboos Univ Med J 2018 May;18(2):e236-e238.
- 5. Shahbahrami A, Pham TA, Bertels K. Parallel implementation of gray level co-occurrence matrices and Haralick texture features on cell architecture. J Supercomput 2012;59(3):1455-1477.
- 6. Ferreira Francisco FA, Pereira e Silva JL, Hochhegger B, Zanetti G, Marchiori E. Pulmonary alveolar microlithiasis. State-of-the-art review. Respir Med 2013 Jan;107(1):1-9.
- 7. Yin X, Wang H, Wu D, Zhao G, Shao J, Dai Y. SLC34A2 Gene mutation of pulmonary alveolar microlithiasis: report of four cases and review of literatures. Respir Med 2013 Feb;107(2):217-222.
- 8. Saito A, McCormack FX. Pulmonary alveolar microlithiasis. Clin Chest Med 2016 Sep;37(3):441-448.
- 9. Ahmad K, Kumar Gupta M, Dhungel K, Lal Sah P, Ansari S, Kumar Rauniyar R. Pulmonary alveolar microlithiasis: a rare case report. Iran J Med Sci 2013 Sep;38(3):267-270.
- 10. Tachibana T, Hagiwara K, Johkoh T. Pulmonary alveolar microlithiasis: review and management. Curr Opin Pulm Med 2009 Sep;15(5):486-490.
- 11. Qublan HS, Athamneh I, Al-Kaisi NS. Azoospermia associated with testicular and pulmonary microlithiasis. J Diagn Med Sonogr 2003;19(3):192-194.