|
Introduction
The global prevalence of sickle cell disease (SCD) and other data estimated that around 20-25 million individuals worldwide have homozygous SCD; 12-15 million in sub-Saharan Africa.1,2 In the West African country of Nigeria, more than 150,000 children are born with the disease annually and 4 million people are afflicted.2-4 Sickle cell anemia (SCA) is the most common genetic disease and affects approximately 2% of Nigerians.5 The protean clinical manifestation of SCA is mainly due to repeated vaso-occlusion chronic intravascular hemolysis, microvascular ischemia and organ damage.4 Splenic complications amongst others are common as a consequence of progressive injury resulting from repeated sickling of sickle hemoglobin in red blood cells. In early life, splenomegaly has been known to be common. The spleen is dark purple and soft with a smooth surface, it has a thin capsule and a thin inconspicuous trabeculae.6,7 Microscopy at this stage revealed congestion of the reticular spaces with sickled erythrocytes and dilatation of the capillaries.7 In later years, however, "autosplenectomy" commonly occurs, the spleen becomes firm, smaller and nodular with depressed scars and finally reduced into a small wrinkled remnant often buried in adhesions.7
Clinically, patients may be asymptomatic accruing gradual damage, or more commonly present with repeated episodes of acute left upper quadrant pain due to splenic infarction.7 Functional asplenia has been reported and is defined as impaired splenic reticulo-endothelial function in spite of splenic enlargement.7 Functional asplenia is associated with increased proneness to infection and may be reversed by blood transfusion.7 The persistence of splenomegaly may be due to endemic malaria.7 Another splenic abnormality is calcification, which is usually demonstrated by radiography in adults.7 There are no specific symptoms, and calcification is usually an accidental finding during radiographic investigation.7
The objective of this study was to determine and review the prevalence of autosplenectomy among patients with sickle cell anemia in a tertiary healthcare facility. Medical advances in the management of sickle cell disease have led to significant increases in the life expectancy of this group of patients. The improvement in general public health education, parental and patient education, improved nutritional care, as well as nutritional supplementation (folic acid, ascorbic acid, pyridoxine), and the advances in red cell transfusion therapy (Top up packed red cell transfusion, exchange transfusion) appear to contribute and influence the clinical course of sickle cell anemia. Also, malarial prophylaxis and other control measures such as penicillin prophylaxis for children, pneumococcal immunization (infection control) and more recently, the institution of Hydroxyurea therapy to induce the increase in foetal hemoglobin have contributed to reducing the chronic hemolytic and vaso-occlusive episodes; consequently reducing splenic infarction and therefore reducing autosplenectomy. Data derived from this study will contribute in optimizing our standard of care as well as initiate a program for the prevention of sickle cell disorders in our locality.
Methods
The study was approved by the Ethical and Scientific Committee of Ahmadu Bello University, Zaria, Nigeria. The study was carried out between May and July 2010. The research subjects were adult Nigerians, 10 years and above of both genders who volunteered and provided written informed consent. Patients were recruited from the hematology clinic. The exclusion criteria included subjects who were unable to understand the investigational nature of the study and unable to give informed consent. Eighty-four sickle cell disease subjects (46 females and 38 males) and 20 controls (10 females and 10 males) were recruited into the study. Subjects underwent complete history, physical examination and laboratory investigations. On electrophoresis, all patients with SCD showed no HbA (but showed elevated levels of HbS), consistent with HbSS phenotype; while the control subjects showed no HbS, consistent with HbAA. Hematological parameters were determined by multi-parameter analyzer Sysmex XT 2000i.
All patients were scanned by B-mode Ultrasonography with Aloka product (model SSD-3500) Ultrasound diagnostic equipment with a variable frequency probe at 2-5MHz or 5-10MHz. Examination was performed with the patient in the supine, right side, or left side position to obtain an optimal view. Measurements of the spleen were performed in all patients and controls along the long axis of the spleen at the level of the hilum to determine its craniocaudal length. Autosplenectomy was defined as the non visualization of the spleen; while Splenomegaly was defined when the long axis of the organ is longer than 130 mm.8 Splenomegaly was defined as "mild" if the two measurements exceeded the upper limit of normal by less than 10%; "moderate" when the measurement was 10% to 20% greater; and "marked" when the measurement exceeded the upper limit of normal by more than 20%.9 A shrunken spleen was defined as the long axis of the spleen measured less than 50 mm.9
Results
Eighty-two consecutive SCA subjects at steady state and 20 healthy non-matched controls were studied in Zaria, Nigeria. However, only 74 of the study subjects had complete data which could be analyzed (one relocated, he only had clinical assessment and no hematologic nor ultrasonographic evaluation, the other two patients did not appear for hematologic/ultrasonographic evaluation and did not respond to phone contact; while the remaining 5 patients had lost/missing data). Data was analyzed with anaylse-it Microsoft Excel. The age range of the study subjects was 10-52 years, while that of the control group was 20-24 years. The mean age of the sickle cell patients was 23.2 ±5.3 years, while that of the control group was 22.7 ±12.4 years. Of the 74 sickle cell subjects, 41 (55.4%) were females; while of the 20 controls, 10 (50%) were females. None of the study subjects had a previous history of a surgical splenectomy. All the study subjects were hemoglobin SS homozygotes (SCA), while all the controls were hemoglobin AA hemozygotes. There was no history of previous blood transfusion in the control subjects. The clinical characteristics of the patients and the controls are presented in Table 1.
Patients with SCD (study subjects) had significantly lower values of hematocrit (25.1% ± 3.9) compared to 40.9% ± 4.6 (p<0.0001) in the controls (Hb AA). The total white blood cell counts were significantly higher in the study group compared to the control group, (p<0.0001). The mean splenic size of the study subjects was 3.5cm ± 4.5, which was also significantly lower than the mean splenic size of the control group (8.7 ± 0.95; I<0.0001). (Table 2)
Table 1: Demographic and Hematological parameters.
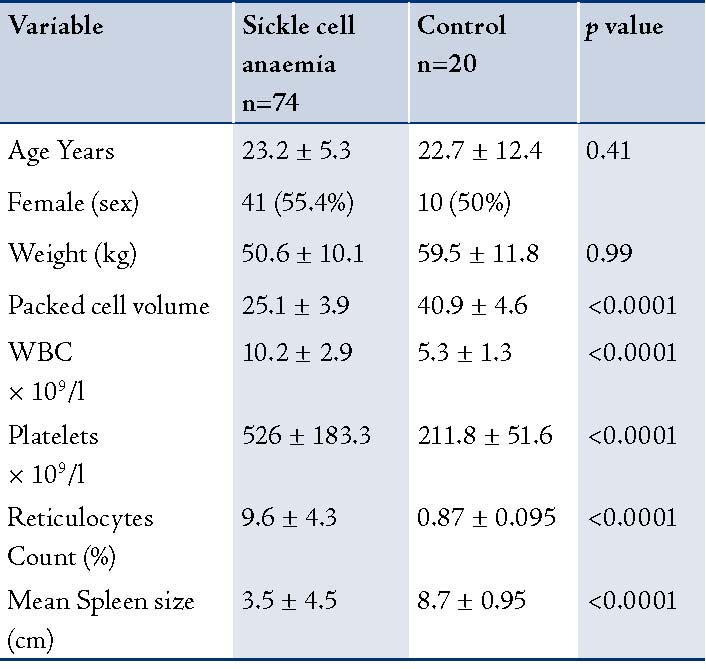
Table 2: Splenic size in the study subjects.

Discussion
In the current study, 41 (55.4%) of the study subjects had autosplenectomy as determined by non-visualization of the spleen during ultrasonographic examination. Although in 23 (31%) patients the spleen was present; it was however, shrunken. This is consistent to the report by Bakhieta Ibrahim Attalla, where autosplenectomy was detected in 43 (47.8%) patients.10 Similarly, it was also established in reports from Jamaica and the US, where autosplenectomy was found in most adult patients with SCD.11,12 However, autosplenectomy was higher in our study compared to the reports from Saudi Arabia by Alsalem et al. where out of the 363 studied patients, only 24 (6.6%) of the patients had autosplenectomy.13 This may be due to the inclusion of subjects older than 10 years in our study or the presence of a less severe Arabian-Indian haplotype in their area of study relatively to the benign haplotype in Nigeria. Our finding of 41% autosplenectomy is also higher than the 33.3% reported by Balci in the study of 102 patients with sickle cell disease at the Center of Hemoglobinopathy of the Antakya State Hospital, Turkey.14
On the other hand, while our study established the general belief of autosplenectomy among sickle cell anemia patients; this conflicts with earlier reports by Olantunji et al. at the University teaching hospital of Ilorin, who did not confirm anatomical autosplenectomy in their study of 98 patients with sickle cell anemia.15 Autosplenectomy is most frequently encountered in patients with homozygous SCD, although it has also been reported in pneumococcal septicemia and systemic lupus erythromatosis.16 Marked splenomegaly of only 2.7% was recorded in the current study; however, the mere presence of splenomegaly in these patients does not imply function (functional asplenia).
This study thus confirms the generally held view of autosplenectomy in sickle cell anemic patients. We therefore advocate the following: (a) an intensified public, patient and parent health education, (b) observation and avoidance of risk/precipitating factors, (c) institution of pneumococcal prophylaxis and vaccination, and (d) availability of effective anti-malarial, antibiotic, nutritional supplements, transfusion support as well as the use of newer therapeutic agents such as Hydroxyurea in the management of this group of patients. This will contribute significantly in reducing the frequency of sickling as well as the degree of autosplenectomy. The limitation of this study is basically the inability to test for splenic function due to the lack of such facilities.
Conclusion
This study substantiates the notion that anatomical autosplenectomy is a common finding in Sickle cell anemic patients. This may be related to inadequate clinical care due to the lack of good health education, ignorance, poverty, or poor standard of care, as well as the lack of newer therapeutic agents. Further studies are needed to determine the association of autosplenectomy with the age of the patient, sickle cell haplotype, clinical manifestation of the disease and its effect on the hematological parameters.
Acknowledgements
The authors reported no conflict of interest and no funding was received on this work.
References
1. Serjeant GR. The case for dedicated sickle cell centers. Ind J Hum Genet 2006;12:148-151 .
2. http://www.who.int/genomics/public/Maphaemoglobin.pdf.
3. Aliyu ZY, Kato GJ, Taylor JV VI, Babadoko A, Mamman AI, Gordeuk VR, et al. Sickle cell disease and pulmonary hypertension in Africa: a global perspective and review of epidemiology, pathophysiology, and management. Am J Hematol 2008 Jan;83(1):63-70.
4. Aliyu ZY, Tumblin AR, Kato GJ. Current therapy of sickle cell disease. Haematologica 2006 Jan;91(1):7-10.
5. Akinyanju OO. A profile of sickle cell disease in Nigeria. Ann N Y Acad Sci 1989;565:126-136.
6. Konotey-Ahulu FI. The sickle cell disease patient. England: Tetteh-A’domeno Publishers, 1996:277-291.
7. Graham R. Sergeant, Beryi E. Sergeant. Sickle cell disease. Great Britain: TJ International Ltd publishers, 3rd ed. 2001:148-169.
8. Dick R, Watkinson A. The liver and spleen. In: Sutton D, ed. Textbook of radiology and imaging. 7th ed. New York: Elsevier, 2002; 737-786.
9. Walker TM, Serjeant GR. Focal echogenic lesions in the spleen in sickle cell disease. Clin Radiol 1993 Feb;47(2):114-116.
10. Bakhieta Ibrahim Attalla. Sonographic Findings in Sudanese Children with Sickle Cell Anemia. Journal of diagnostic medical sonography; vol. 26 no 6: 276-280.
11. Watson RJ, Lichtman HC, Shapiro HD. Splenomegaly in sickle cell anemia. Am J Med 1956 Feb;20(2):196-206.
12. Rossi EC, Westring DW, Santos AS, Gutierrez J. Hypersplenism in sickle cell anemia. Arch Intern Med 1964 Sep;114:408-412.
13. Al-Salem AH, Al-Aithan S, Bhamidipati P, Al-Jam’a A, Al Dabbous I. Sonographic assessment of spleen size in Saudi patients with sickle cell disease. Ann Saudi Med 1998 May-Jun;18(3):217-220.
14. Balcı A, Karazincir S, Sangün O, Gali E, Daplan T, Cingiz C, et al. Prevalence of abdominal ultrasonographic abnormalities in patients with sickle cell disease. Diagn Interv Radiol 2008 Sep;14(3):133-137.
15. Olantunji AA, Olatunji PO. Splenic size determination in sickle cell anaemia: An ultrasonographic study. East African Journal 2001;78(7):365-370.
16. Uthman I, Soucy JP, Nicolet V, Senécal JL. Autosplenectomy in systemic lupus erythematosus. J Rheumatol 1996 Oct;23(10):1806-1810.
|