|
Abstract
Subjects with Familial hypercholesterolemia are at increased risk for cardiac events such as premature myocardial infarction and early death from coronary heart disease, especially in patients with severe forms of the disease if left unattended. Therefore, there is an ardent need for the early diagnosis followed by aggressive therapeutic intervention and lifestyle modification. Three groups have developed clinical diagnostic tools for Familial hypercholesterolemia: the US MedPed Program, the Simon Broome Register Group in the United Kingdom, and the Dutch Lipid Clinic Network. In this article, the individual criteria are reviewed with particular emphasis on their advantage(s) and disadvantage(s), and in turn assess their suitability in diagnosing Familial hypercholesterolemia in the Omani Arab population. A brief insight into the process of "Cascade Screening" is also provided, this is a procedure that we are in the process of establishing in Oman.
Keywords: Familial hypercholesterolemia; Simon-Broome Criteria; Cascade-Screening; Dutch Criteria; US-Medped Criteria; Omani Arab Population.
Introduction
Familial hypercholesterolemia (FH) is a genetic disorder of lipid metabolism that is typified by considerable elevations in levels of low-density lipoprotein cholesterol (LDL-C), from which patients are at an elevated risk for premature coronary heart disease (CHD).1,2 The etiology of FH comprises of known mutations in the gene of the LDL receptor (LDLR) (with more than 1600 identified to date,3 the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene, the gene of apolipoprotein B (ApoB) or rare mutations in LDL receptor adapter protein 1 (LDLRAP1) gene.4
Heterozygous FH (HeFH) is the widely occurring form of the disease (prevalence of approximately 1 in 300 to 500 individuals globally and as high as 1 in 100 persons in some populations), with the preponderance of inheritance through an autosomal-dominant mechanism.3-6 Homozygous FH (HoFH) on the other hand, is a very rare form of the disease (prevalence of 1 in 1 million persons).7 However, some populations such as the French-Canadians, Lebanese (both in Brazil and Lebanon), Ashkenazi Jews, and Dutch Afrikaners, are at a significantly increased risk for FH owing to an augmented prevalence of HeFH-associated mutations in the LDLR, in these so called "founder" populations.8-12
Individuals with FH generally have drastically higher levels of LDL-C than most patients with severe hyperlipidemia. Case in point, subjects with FH above the age of 30 years often exhibit an LDL-C level of 250 milligrams per deciliter (mg/dL) (13.9 mmol/L) or higher; patients between 20 and 29 years of age often exhibit LDL-C levels of 220 mg/dL (12.2 mmol/L) or higher; while patients younger than 20 years of age often have an LDL-C level of 190 mg/dL (10.5 mmol/L) or higher.3 Although high cholesterol levels more often than not do not exhibit any physical symptoms, cholesterol may be deposited in specific areas in the body that may be visible from outside. These include, regions around the eyelids (xanthelasma palpebrarum), as a white ring in front of the periphery of the iris (arcus senilis corneae), and in the form of lumps in the tendons of the hands and feet specifically the Achilles tendon (tendon xanthoma).13,14
Subjects with FH are at increased risk for cardiac events such as premature myocardial infarction and early death from CHD, especially in patients with severe forms of the disease if left unattended. Risk factors for CVD in patients with FH are comparable to those in patients without FH, although FH itself is a significant CVD risk factor because it results in long-term exposure to high lipid levels.4,15,16 In HeFH, there is one normally functioning gene and therefore, the disease condition is less severe and more responsive to treatment such as statins than HoFH. The mean age for the inception of CVD in men with HeFH is 42 to 46 years and in women the onset occurs around 51 to 52 years.17,18 HoFH on the other hand, is an aggressive form of FH, which is frequently unresponsive to conventional treatment for hypercholesterolemia owing to the patient’s lack of functional LDLR.16,18 In patients with HoFH, the mean age at the time of diagnosis of CVD is 20 years.15
Therefore, there is an ardent need for early diagnosis followed by aggressive therapeutic intervention and lifestyle modification. The clinical diagnosis of FH is founded on personal and family history, physical examination, and lipid concentrations. Three groups have developed clinical diagnostic tools for FH: the US MedPed Program, the Simon Broome Register Group in the United Kingdom, and the Dutch Lipid Clinic Network.
The question that arises is, "Which of the clinical diagnostic tools is more suitable for the diagnosis of FH in a genotypically homogenous population such as the Omani Arab population and likewise?" Additionally, there are other factors such as cost-effectiveness and ease of applicability which should also be taken into consideration. In this epigrammatic review, the individual clinical diagnostic tools are discussed in order to appraise the suitability of their application to the diagnosis of FH in the Omani Arab population.
The Simon Broome Criteria
The Simon Broome criteria takes into consideration cholesterol concentrations, clinical characteristics, molecular diagnosis, and family history, which include risk of fatal CHD in FH.19 The criteria are summarized in Table 1.
Table 1: The Simon-Broome Diagnostic Criteria.

According to this criterion, definite FH is defined as:
total cholesterol higher than 6.7 mmol/L or LDL-C greater than 4.0 mmol/L in a child whose aged under 16 years or total cholesterol greater than 7.5 mmol/L or LDL-C higher than 4.9 mmol/L in an adult (levels either pre-treatment or highest on treatment).
plus
tendon xanthomas in the patient, or in a first-degree relative (parent, sibling or child), or in a second-degree relative (grandparent, uncle or aunt)
or
DNA-based confirmation of an LDLR mutation, familial defective ApoB-100, or a mutation in the PCSK9 gene.
The Simon Broome Criteria directs that for a diagnosis of possible FH:
total cholesterol should be higher than 6.7 mmol/L or LDL-C should be grater than 4.0 mmol/L in a child whose aged under 16 years or total cholesterol greater than 7.5 mmol/L or LDL-C greater than 4.9 mmol/L in an adult (levels either pre-treatment or highest on treatment)
and at least one of the following
family history of myocardial infarction: younger than 50 years of age in a second-degree relative or younger than 60 years of age in a first-degree relative
or
family history of raised total cholesterol: greater than 7.5 mmol/L in an adult first- or second-degree relative or greater than 6.7 mmol/L in a child or sibling aged younger than 16 years.
Advantage of the Simon-Broome Criteria:
1. Ease of Remembrance: The criteria is set out in such a fashion that it can easily be remembered easily and therefore its applicability in a day-to-day lipid clinic is easy and effective. In fact, of the three criteria currently available, it is the most widely used by physicians.
2. Economic viability: The Simon-Broome criteria at a glance uses the conventional physical signs, history taking and non-invasive procedure of lipid profile analysis for the diagnosis of FH. This as a whole makes the application of this criterion easy and economically feasible. In order to diagnose definite or possible FH, the physician does not have to resort to expensive DNA testing. This makes the suitability of the criteria to be applied in a healthcare center setting such as the type observed in Oman,20 whereby patients diagnosed with definite or possible FH are then referred to major hospitals such as Sultan Qaboos University Hospital later for molecular testing.
Disadvantage of the Simon-Broome Criteria
Simon-Broome criteria fails to differentiate between classical FH due to LDLR mutations and the other genetic origins of FH such as ApoB-100 and PCSK9. It will also not discriminate between FH and non-FH such as secondary hypercholesterolemia, sitosterolemia, or other variants. More importantly, the criteria may overlook a substantial proportion of FH patients, mainly those with a mild phenotype and the pediatric population in whom the phenotype has not yet emerged. In fact, a myocardial infarction is the first presenting sign in many FH patients. Additionally, the criteria does not allocate for understanding of known genotype-phenotype correlations such as better response to statin therapy in ApoB-100 and ARH compared to LDLR mutations.21
The Dutch Criteria
The Dutch criteria for the diagnosis of FH are the modification of the Simon-Broome criteria. The principal reason for developing the Dutch criteria is that the Simon-Broome criteria diagnoses FH based on personal and family history, physical examination, and laboratory findings. However, the underlying molecular defect of FH is overlooked in the Simon-Broome criteria. To address this shortcoming, the Dutch criteria introduce a point system and take into consideration the molecular defect of FH. The criteria are represented in Table 2.
Table 2: The Dutch Diagnostic Criteria.

In the Dutch criteria,22 points take into consideration family history of hyperlipidemia or heart disease, clinical characteristics such as tendinous xanthomata, elevated LDL cholesterol, and/or an identified mutation. A total point score of greater than 8 is considered "definite" FH, 6–8 is "probable" FH, and 3–5 is "possible" FH. In comparison to the Simon Broome, the Dutch criteria requires that at least one other criterion be met in addition to molecular diagnosis.22
One of the key advantages of the Dutch criteria is that it addresses the molecular defect that leads to FH and this in turn will help to provide the diagnosed FH patient with a better therapeutic strategy/treatment.
Disadvantages of the Dutch Criteria
1. Lack of versatility of use: Unlike the Simon Broome criteria, the Dutch criteria lacks versatility. Firstly, its use in the day-to-day clinic is cumbersome as for a definite diagnosis of FH points obtained for the patient should be 8 or higher. Secondly, the chart corresponding to the criteria if adequately adhered to will require entailing a longer time of attendance by the physician for a given patient. This means that if there is a larger number of patients attending the clinic on a given day, the diagnosis of FH may be erroneous due to stress over time that the physician may be exposed to. Additionally, attendance of lipid clinics by patients is often governed by demographics, clinical parameters and treatment beliefs. In a prospective cohort study by Elis et al. it was observed that beliefs involving lipid-lowering treatment should be effectively identified, so that they may be elucidated in order to improve patient attendance at follow-up visits to a lipid clinic.23 Therefore, if the Simon-Broome Criteria is compared vis-‡-vis with the Dutch criteria, offering suitable details to the patient with regards to the criteria for diagnosis of FH is relatively easier for the Simon-Broome criteria as it offers a straight forward scheme of diagnosis in comparison to the Dutch criteria. Among the Omani population, visits to a health professional are often governed by cultural and traditional beliefs.24 Therefore, putting forth a simplistic explanation with regard to diagnostic criteria pertaining to FH may make a difference between a patients revisit to the clinic for proper treatment and management of FH. In such a case, the clinician may find that providing a suitable elucidation is relatively easier with regards to the Simon Broome criteria.
2. Expensive: One of the key points in the Dutch Criteria is the elucidation of molecular defect for the diagnosis of FH. This requires that the genomic DNA for FH associated genes such as LDLR, PCSK9 and ApoB-100 be screened in order to identify the defect. For such a screening process, a DNA sequencer and sequencing reagents will be required, which are both expensive (as explained in Fig. 1) and difficult to be applied in a health center setting (such as those established in Oman) as handling such equipment requires suitably trained personnel. As observed in Fig. 1, the cost of sequencing a megabase of DNA has fallen over the decade and has defied even the Moore’s law (the Law predicts that every two years the cost of computing will fall by half.25 In other words, in the years to come, gadgets (especially involving electronic circuitry) will be better as well as cheaper. In terms of DNA sequencing with the evolution of the DNA sequencer in terms of the electronics involved, the cost of sequencing will go down. However, in case of FH, one needs to screen three genes which will still entail a substantial fee per patient. Additionally in recent studies, it has been shown that additional genes apart from that of LDLR, PCSK9 and ApoB-100 may be involved in causing FH,21 as a result of which the whole genome or at least the exome needs to be screened for alteration in order to be definitive.
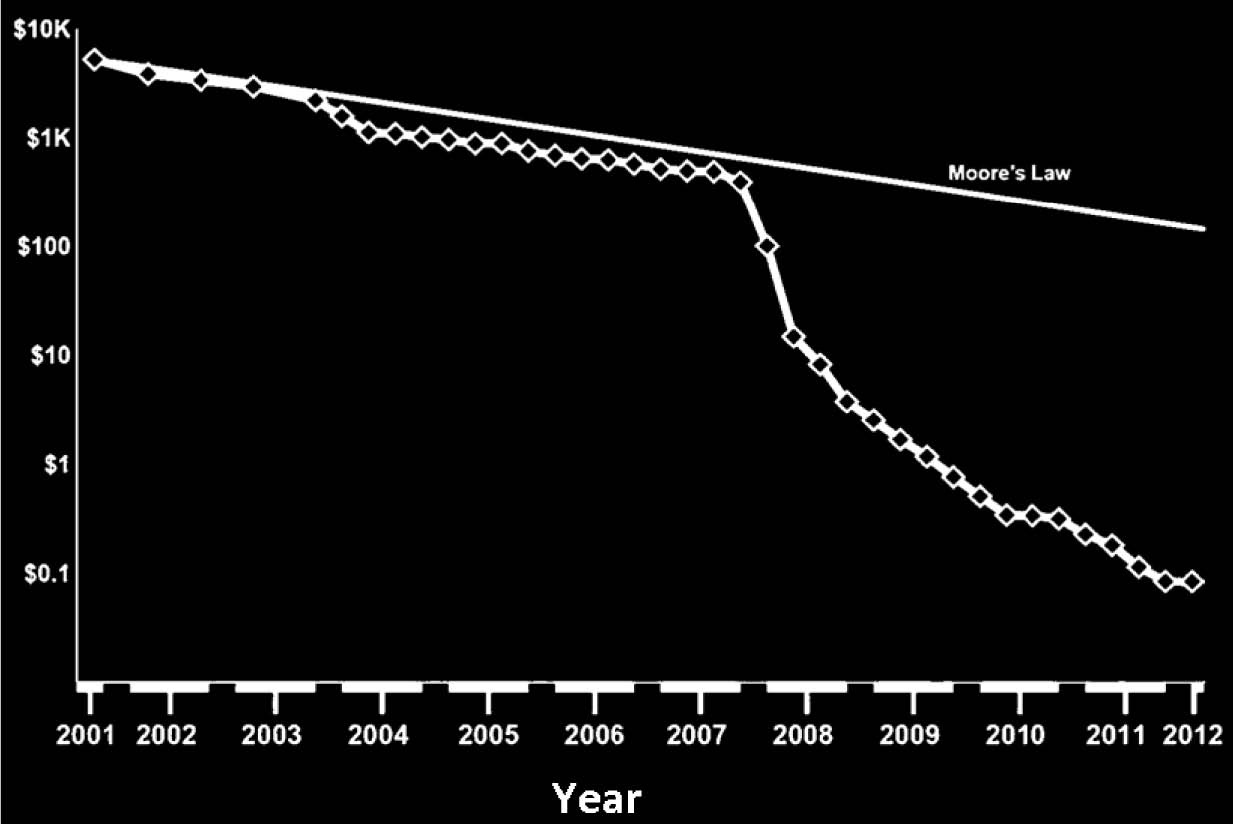
Figure 1: Association between cost of sequencing a Megabase of DNA and Technical Advancement over the last Decade. The cost of sequencing a megabase of DNA with the advent of new generation sequencers have fallen over the last decade and has even defied Moore’s law (solid line in the figure, [data represented from 25]).
The US MEDPED Criteria
The US MEDPED criteria uses age-specific and relative specific criteria for total cholesterol only (Table 3).26 A point to note regarding these criteria is to be aware of the lower total cholesterol diagnostic cut-off levels in patients with first-, second- or third-degree relatives with FH compared with the cut-off levels for the general population. (Table 3)
Table 3: The US (MEDPED) diagnostic criteria for FH.
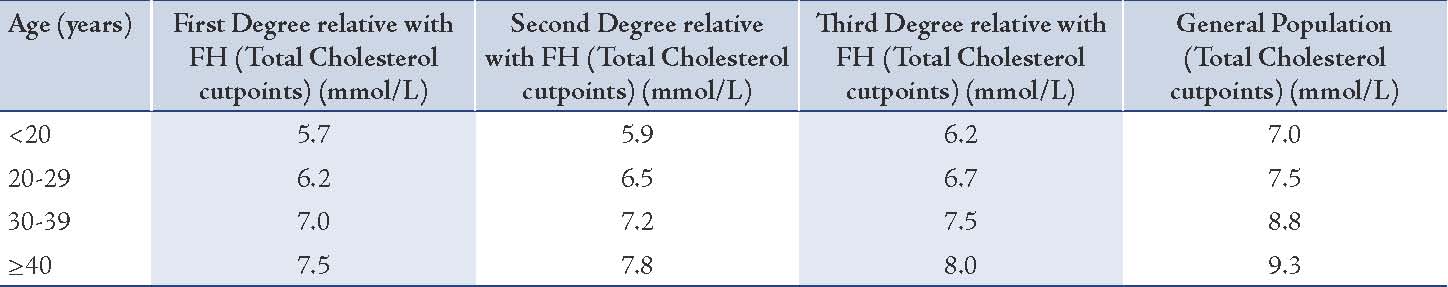
Disadvantage of the US MEDPED Criteria
Although easy to use, the criteria does not take into consideration the clinical characteristics such as tendinous xanthomata and/or an identified mutation in the FH-associated gene although it advocates cascade screening (refer below) once FH has been diagnosed by employing the criteria. Additionally, various factors influence the variation in the levels of total cholesterol. Case in point, in a longitudinal study of seasonal variation in lipid levels in 517 healthy volunteers from a health maintenance organization serving central Massacheussetts; it was observed over a 12-month period that a seasonal variation existed in the levels of lipids where the variation is higher in women and hypercholesterolemic individuals, with alterations in plasma volume accounting for much of the disparity. A relative plasma hypervolemia during the summer also seems to be linked with increases in temperature and/or physical activity.27
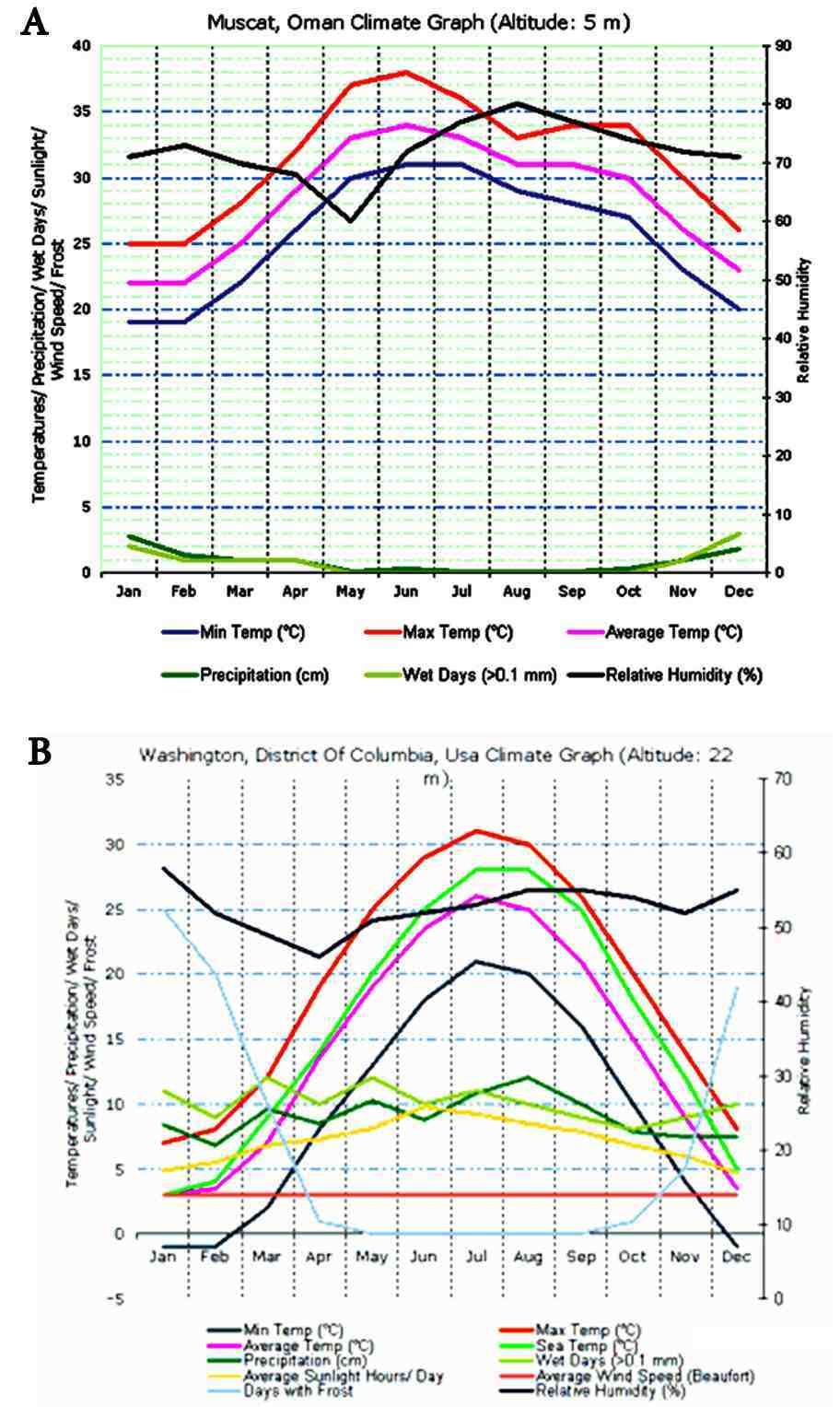
Figure 2: Comparison of Temperatures between Muscat and Washington DC throughout the year. Muscat (A), experiences predominant summer temperatures compared with Washington DC (B) and therefore in line with the studies by Ockene et al.27 the population of Muscat will exhibit significantly higher levels of lipids compared to the population in Washington DC, making the use of US MEDPED criteria difficult in Oman.
In order to determine how temperature variations my affect limit levels, the year-round temperature for Muscat, Oman was plotted and compared with that of Washington DC, USA (Fig. 2) using the online temperature plot module of http://www.climatemps.com. As observed in Fig. 2, Muscat experiences predominantly summer weather all year-round whereas in Washington, the temperatures are far cooler. Therefore, in line with the observations of Ockene et al.27 the population in Muscat will plausibly tend to exhibit higher lipid levels in comparison to the levels observed in the US. Therefore, the US MEDPED criteria, which solely depends on the diagnosis of FH based on the values of total cholesterol, may not be applicable to the Omani Arab population.
Diagnosis of FH in Oman
Recently, two cases of FH were reported from two different Omani Arab families.7,22 In both cases, the Simon-Broome criteria was applied for diagnosing FH and it was confirmed later through DNA analysis that in one of the families, FH was due to mutation in the LDLR but not in the PCSK9 or the ApoB genes.7 Whereas in the other family FH was due to mutations in both LDLR and PCSK9 genes.21 Both of these studies indicate towards the fact that the Simon-Broome criteria can be successfully applied for the diagnosis of FH in the Omani Arab population.
Cascade Screening
In 1997, the World Health Organization (WHO) evidently recognized the benefits of a DNA test for the diagnosis of FH and re-assured that it was cost-effective to do so.28 However, with the evolving polygenetic nature of FH, several studies have demonstrated that genetic diagnosis is hindered by the towering cost, and genetic screening for the population at large failed to show cost-effectiveness levied to the polygenetic nature of FH.
However, the recent advent of the cascade-screening strategy may solve this issue of cost-effectively. In cascade screening, an index patient is initially diagnosed clinically by employing one of the clinical criteria listed in Tables 1 to 3. Following which a DNA test is performed, the DNA test confirms the mutation in the index patient. Screening for the matching mutation is commenced in first-degree relatives to look for fresh cases of FH. New inveterate cases from the relatives are treated as new index cases and their first-degree relatives are screened. The concept of cascade screening has been successfully applied in the Dutch,29 Norwegian,30 Spanish,31 Australian,32 Wales,19 and the population in New Zealand.32 A similar strategy can be applied to the genetically homogenous Omani Arab population following the diagnosis of FH using the Simon-Broome criteria.
Exome Sequencing Strategy
Exome sequencing or the targeted sequencing of the subset of the human genome that is protein-coding — is an authoritative and cost-effective newfangled tool for dissecting the genetic basis of diseases and traits that have ascertained to be intractable to conventional gene-discovery strategies.33 Over the past years, experimental and analytical approaches relating to exome sequencing have proven to be a rich framework for ascertaining the genes underlying unresolved Mendelian disorders. Additionally, exome sequencing is being adapted to explore the extent to which rare alleles explain the heritability of complex diseases and health-related traits. Although in FH, mutations in three genes are associated with causing the disease, a complex genetic etiology may exist, as these genetic loci may interact with other unidentified loci indicating towards the degree of severity of the disease.
Case in point, exome sequencing in a patient suspected with mitochondrial disease revealed a homozygous splice site mutation in SETX, which is known to cause Spinocerebellar Ataxia, Autosomal Recessive 1 (SCAR1). Further, a missense mutation was also identified in a highly conserved position of the OCRL gene, which causes Lowe Syndrome and Dent Disease 2.34 On reviewing this patient's complex phenotype, it was observed that a complex genetic etiology was present in which no single gene explained the complete clinical presentation. Thus, exome sequencing reveals that this patient did not have mitochondrial disease but rather a genocopy caused by more than one mutant locus. Therefore, in the distant future, mandated sequencing of the exome for patients with severe presentation of FH may help towards better management of the disease in addition to the elucidation of the "interactome" that may exist between the already identified genes with the ones that are still to be identified.
Conclusion
Overall, the Simon-Broome Criteria is most suitable for diagnosing FH in the Omani Arab population, although it does not diagnose the condition at the genetic level. Following diagnosis using the Simon Broome, a cascade screening strategy can be availed to pursue genetic testing of FH. A project is currently being designed to appraise the cascade screening strategy in the Omani Arab population. With the establishment of a cascade screening strategy, further investigations can be carried out to promote the exome sequencing approach in order to improve our understanding and management of the disease.
Acknowledgements
The authors reported no conflict of interests and no funding was received for this work.
References
1. Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis 2012 Aug;223(2):262-268.
2. Jensen HK. The molecular genetic basis and diagnosis of familial hypercholesterolemia in Denmark. Dan Med Bull 2002 Nov;49(4):318-345.
3. Hopkins PN, Toth PP, Ballantyne CM, Rader DJ; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Familial hypercholesterolemias: prevalence, genetics, diagnosis and screening recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol 2011 Jun;5(3)(Suppl):S9-S17.
4. Liyanage KE, Burnett JR, Hooper AJ, van Bockxmeer FM. Familial hypercholesterolemia: epidemiology, Neolithic origins and modern geographic distribution. Crit Rev Clin Lab Sci 2011 Jan-Feb;48(1):1-18.
5. Goldberg AC, Hopkins PN, Toth PP, Ballantyne CM, Rader DJ, Robinson JG, et al; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol 2011 Jun;5(3)(Suppl):S1-S8.
6. Wierzbicki AS, Humphries SE, Minhas R; Guideline Development Group. Familial hypercholesterolaemia: summary of NICE guidance. BMJ 2008;337:a1095.
7. Al-Hinai AT, Al-Abri A, Al-Dhuhli H, Al-Waili K, Al-Sabti H, Al-Yaarubi S, et al. First case report of familial hypercholesterolemia in an Omani family due to novel mutation in the low-density lipoprotein receptor gene. Angiology 2013 May;64(4):287-292.
8. Hobbs HH, Brown MS, Russell DW, Davignon J, Goldstein JL. Deletion in the gene for the low-density-lipoprotein receptor in a majority of French Canadians with familial hypercholesterolemia. N Engl J Med 1987 Sep;317(12):734-737.
9. Moorjani S, Roy M, Gagné C, Davignon J, Brun D, Toussaint M, et al. Homozygous familial hypercholesterolemia among French Canadians in Québec Province. Arteriosclerosis 1989 Mar-Apr;9(2):211-216.
10. Vohl MC, Moorjani S, Roy M, Gaudet D, Torres AL, Minnich A, et al. Geographic distribution of French-Canadian low-density lipoprotein receptor gene mutations in the Province of Quebec. Clin Genet 1997 Jul;52(1):1-6.
11. Yuan G, Wang J, Hegele RA. Heterozygous familial hypercholesterolemia: an underrecognized cause of early cardiovascular disease. CMAJ 2006 Apr;174(8):1124-1129.
12. Defesche JC, van Diermen DE, Lansberg PJ, Lamping RJ, Reymer PW, Hayden MR, et al. South African founder mutations in the low-density lipoprotein receptor gene causing familial hypercholesterolemia in the Dutch population. Hum Genet 1993 Dec;92(6):567-570.
13. Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest 2003 Jun;111(12):1795-1803.
14. Tsouli SG, Kiortsis DN, Argyropoulou MI, Mikhailidis DP, Elisaf MS. Pathogenesis, detection and treatment of Achilles tendon xanthomas. Eur J Clin Invest 2005 Apr;35(4):236-244.
15. Kolansky DM, Cuchel M, Clark BJ, Paridon S, McCrindle BW, Wiegers SE, et al. Longitudinal evaluation and assessment of cardiovascular disease in patients with homozygous familial hypercholesterolemia. Am J Cardiol 2008 Dec;102(11):1438-1443.
16. Naoumova RP, Thompson GR, Soutar AK. Current management of severe homozygous hypercholesterolaemias. Curr Opin Lipidol 2004 Aug;15(4):413-422.
17. Alonso R, Mata N, Castillo S, Fuentes F, Saenz P, Muñiz O, et al; Spanish Familial Hypercholesterolaemia Group. Cardiovascular disease in familial hypercholesterolaemia: influence of low-density lipoprotein receptor mutation type and classic risk factors. Atherosclerosis 2008 Oct;200(2):315-321.
18. Jansen AC, van Aalst-Cohen ES, Tanck MW, Trip MD, Lansberg PJ, Liem AH, et al. The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: data in 2400 patients. J Intern Med 2004 Dec;256(6):482-490.
19. Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ 1991 Oct;303(6807):893-896.
20. Dhar A. Medical informatics and health care in Oman. J Am Med Inform Assoc 2001 Sep-Oct;8(5):515-517.
21. Al-Waili K, Al-Zidi WA, Al-Abri AR, Al-Rasadi K, Al-Sabti HA, Shah K, et al. Mutation in the PCSK9 Gene in Omani Arab Subjects with Autosomal Dominant Hypercholesterolemia and its Effect on PCSK9 Protein Structure. Oman Med J 2013 Jan;28(1):48-52.
22. Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol 2004 Sep;160(5):407-420.
23. Elis A, Lishner M, Melamed S. Treatment beliefs and attending follow-up visits in a lipid clinic. Br J Health Psychol 2011 Feb;16(Pt 1):61-71.
24. Al-Busaidi ZQ. A Qualitative Study on the Attitudes and Beliefs towards Help Seeking for Emotional Distress in Omani Women and Omani General Practitioners: Implications for Post-Graduate Training. Oman Med J 2010 Jul;25(3):190-198.
25. Martín J, Cervero A, Mir P, Martinez-Conejero JA, Pellicer A, Simón C. The impact of next-generation sequencing technology on preimplantation genetic diagnosis and screening. Fertil Steril 2013 Mar;99(4):1054-1061, e3.
26. Williams RR, Hunt SC, Schumacher MC, Hegele RA, Leppert MF, Ludwig EH, et al. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol 1993 Jul;72(2):171-176.
27. Ockene IS, Chiriboga DE, Stanek EJ III, Harmatz MG, Nicolosi R, Saperia G, et al. Seasonal variation in serum cholesterol levels: treatment implications and possible mechanisms. Arch Intern Med 2004 Apr;164(8):863-870.
28. Wonderling D, Umans-Eckenhausen MA, Marks D, Defesche JC, Kastelein JJ, Thorogood M. Cost-effectiveness analysis of the genetic screening program for familial hypercholesterolemia in The Netherlands. Semin Vasc Med 2004 Feb;4(1):97-104.
29. Umans-Eckenhausen MA, Defesche JC, Sijbrands EJ, Scheerder RL, Kastelein JJ. Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands. Lancet 2001 Jan;357(9251):165-168.
30. Leren TP, Manshaus T, Skovholt U, Skodje T, Nossen IE, Teie C, et al. Application of molecular genetics for diagnosing familial hypercholesterolemia in Norway: results from a family-based screening program. Semin Vasc Med 2004 Feb;4(1):75-85.
31. Pocovi M, Civeira F, Alonso R, Mata P. Familial hypercholesterolemia in Spain: case-finding program, clinical and genetic aspects. Semin Vasc Med 2004 Feb;4(1):67-74.
32. Muir LA, George PM, Laurie AD, Reid N, Whitehead L. Preventing cardiovascular disease: a review of the effectiveness of identifying the people with familial hypercholesterolaemia in New Zealand. N Z Med J 2010 Nov;123(1326):97-102.
33. Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009 Sep;461(7261):272-276.
34. Craigen WJ, Graham BH, Wong LJ, Scaglia F, Lewis RA, Bonnen PE. Exome sequencing of a patient with suspected mitochondrial disease reveals a likely multigenic etiology. BMC Med Genet 2013;14:83.
|