|
|
Abstract
Microvillous Inclusion Disease (MVID) is one of the congenital diarrheal disorders (CDD) caused by genetic defects in enterocyte differentiation and polarization. Its prevalence is higher in countries with a high degree of consanguinity. It causes severe, intractable secretory diarrhea leading to permanent and definitive intestinal failure with resultant dependency on parenteral nutrition (PN). Small bowel transplantation is the only curative treatment. The gold standard for diagnosis are the typical morphological abnormalities in small bowel biopsies on light and electron microscopy (EM). In recent times, histochemistry and immunohistochemistry haves hown sufficient diagnostic accuracy replacing EM if the facility is unavailable or EM findings are inconclusive. We describe a neonate with CDD who was diagnosed to have MVID on the duodenal biopsy by morphohistochemical and immunophenotypic methods used for the first time in Oman. By utilizing such easy and accessible diagnostic methods, a rare genetic disorder could be diagnosed with certainty and the family could be counseled accordingly. With a high degree of consanguinity in the region, the prevalence of MVID in Oman needs to be identified once these patients are diagnosed by utilizing appropriate investigations. Care of such patients necessitates improving current parenteral nutrition services and addressing the future need for small bowel transplantation (SBTx), in Oman.
Keywords: Congenital diarrheal disorder; Microvillous Inclusion Disease; Small intestine biopsy; Intestinal transplantation.
Introduction
Congenital diarrheal disorders (CDD) are rare enteropathies generally inherited as autosomal recessive traits.1,2 Infants affected usually present with severe diarrhea within a few hours, days or months of life leading to life-threatening dehydration and metabolic acidosis that requires PN.2,3 CDDs are challenging conditions because of their severity and the broad differential diagnosis.2,3 MVID (OMIM 251850) is one of the CDDs resulting from defects of enterocyte differentiation and polarization. Its prevalence is higher in countries with a high degree of consanguinity.4 Children with MVID develop marked diarrhea, dehydration and metabolic acidosis. They are at risk of developing cholestasis and liver failure secondary to PN. Endoscopic examination of the small bowel (SB) and colon is usually normal. Diagnosis has traditionally rested on typical morphological abnormalities detected through light and EM examinations of SB biopsies.5 In the past decade, histochemical,6 and more recently immunohistochemical stains,7 have supplanted EM as simple and easily available diagnostic tools with a sensitivity and specificity matching EM. The latter is available at fewer centers and its usefulness rests on sampling.
This is a report of an infant diagnosed with MVID, based on a clinical suspicion supported by light microscopic features and pattern of histochemical (Periodic Acid Schiff - PAS) and immunohistochemical (CD 10) stains on SB biopsy. EM features were complementary to the final diagnosis. This report highlights the significance of this disease to Oman in particular. The use of PAS and CD10 stains to diagnose MVID is reported for the first time in Oman. It made the diagnosis easier, practical and specific. In future, this will help to accurately determine MVID prevalence in the country.
Case Report
A term female born via spontaneous vaginal delivery with no complications. Parents are consanguineous with one healthy child. She was well and was breast-fed for 3 days of life when she started to have frequent large watery stools 10 times per day. There was no blood or mucous. She soon developed lethargy and poor feeding with 25% weight loss. She was seen at the local hospital and managed with intravenous fluids (IVF) and was treated as neonatal sepsis. She was kept Nil Per Os (NPO) for 3 days with mild reduction in stool output that worsened with feeding reintroduction. She was transferred at day 11 of life to a tertiary hospital for further management.
On examination, she was severely dehydrated with anuria and irritability. There were no dysmorphic features. Abdomen was soft and not distended. Systemic examination was unremarkable. The impression of congenital diarrheal disorder was addressed. Blood investigations showed prerenal azotemia with high urea and creatinine and hyperchloremic metabolic acidosis with high anion gap. She had a picture of proximal tubular dysfunction, likely due to either acute tubular necrosis secondary to prerenal azotemia or congenital Fanconi syndrome. Serum sodium was normal. Serum ammonia and lactate were high (197 umol/L and 4.46 mmol/L, respectively) due to poor perfusion. She was resuscitated with IVF boluses, kept NPO and started on PN. Diarrhea continued with stools output exceeding 100 ml/kg/day. Differential diagnosis included secretory form of CDD with secondary lactic acidosis and renal tubular acidosis. Evaluation of CDD included negative stool white blood cells, blood, urine, cerebrospinal fluid and stool cultures as well as virology on multiple occasions.
Diagnostic trial of elemental formula for possible Cow’s milk protein intolerance and a formula for glucose-galactose malabsorption was initiated but failed with recurrence of high stool output leading to dehydration and acidosis which ruled out such possibilities. Work up for galactosemia and abetalipoproteinemia was negative. Esophageogastro-duodenoscopy (EGD) showed flat duodenal folds. Microscopically, there was significant atrophy of the villi with attenuation of the brush border (Fig. 1) and vacuolation of the enterocytes. (Fig. 1 Inset)
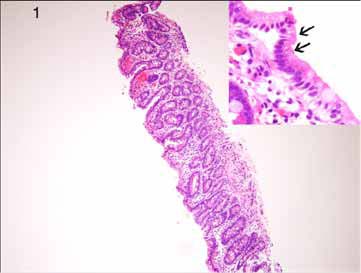
Figure 1: Duodenal biopsy showing ‘thin’ mucosa due to villous atrophy (H&E × 40). Inset: The surface enterocytes show vacuolation (arrows) (H&E × 200).
PAS stain showed an enlarged intracytoplasmic band with PAS positive granules in the apical pole of enterocytes in contrast to the thin uniform linear staining of the brush border in the normal mucosa (Figs. 2A & B). Immunohistochemical staining with CD 10 antibody showed a similar loss of normal linear staining of the brush border (Figs. 2C & D). EM confirmed the atrophic microvilli but the inclusions with abortive microvilli were seen only focally (Figs. 3A & B). The morphohistochemical and immunophenotypic findings were diagnostic of MVID.
The family was extensively counseled regarding the diagnosis and its poor prognosis. The clinical course was complicated by recurrent central line infections with septic shock requiring repeated courses of intravenous broad-spectrum antibiotics and inotropic support. She developed PN liver related disease in the form of cholestasis and liver failure. The family had low socioeconomic and poor educational background that could not allow the child to receive PN at home. SBTx, the definite cure for MVID, being not available in Oman, was not offered to this infant. She was continued on PN at the local hospital. She eventually expired at age of 4 months secondary to recurrent line infections with deteriorating liver disease.
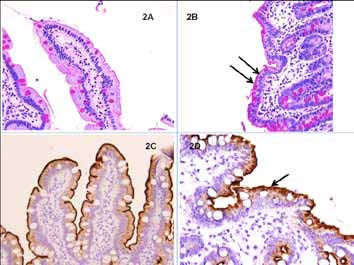
Figure 2: PAS staining of surface enterocytes in the duodenal biopsy. (C&D): CD 10 staining of the brush border. (A): Thin, uniform, linear PAS-stained brush border in normal mucosa. (B): The patient’s mucosal biopsy shows a wide, irregular, intracytoplasmic PAS-positive band with granules at the apical pole of the enterocytes (arrows [Periodic Acid Schiff × 100]). (C): Linear brush border staining in normal mucosa (D): Irregular staining in MVID (CD10 with DAB × 100).
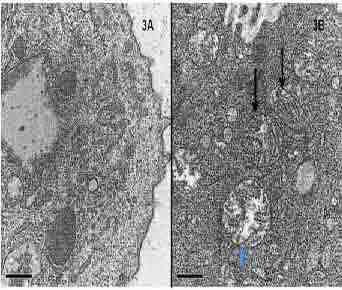
Figure 3: Ultrastructural abnormalities in surface enterocytes. (A): Microvillous atrophy of the surface of enterocytes. (B): Microvillous inclusions (arrows [Uranyl acetate-lead citrate × 4000]).
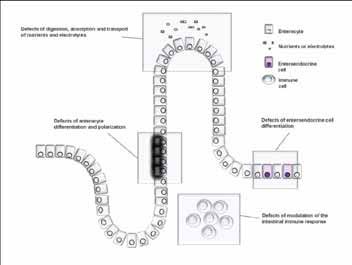
Figure 4: The mechanisms involved in the pathogenesis of congenital diarrheal disorders, Canani et al.3
Discussion
The exact incidence of CDD differs among populations and geographic areas.2 Milder forms remain undiagnosed until adulthood.1,3 CDD includes a large number of conditions. Canani et al.1,3 suggested that CDD could be classified into 4 groups (Fig. 4). New genes have been identified in relation to CDD.3 MVID is one of the CDD according to this classification.1,3 No prevalence data are available. Almost half of 24 patients with MVID in one report were from consanguineous families from the Mediterranean area.8 Rab8, a small GTP-binding protein, and myosin Vb (MYO5B) were demonstrated to be involved in the intracellular transport of proteins to the apical level of the intestinal epithelial cells.9 Several mutations in MYO5B and loss of MYO5B function in MVID patients have been described recently.9-11 MYO5B deficiency may block the apical traffic of intracellular vacuoles containing microvilli, thereby determining aggregation of apically bound vesicles.9
Pregnancy and delivery are usually uneventful. Polyhydramnios is reported in rare isolated cases.4, 12 It causes severe and intractable secretory diarrhea (150-300 ml/kg/day) that usually starts shortly after birth and persists despite total bowel rest,1,2,4,8,13 resulting in severe dehydration and profound metabolic acidosis.2,8-10 A late onset form (3-4 months of life) with a better outcome is described.2 Our patient had a typical presentation. Complete and prolonged bowel rest allows reducing stool volume, but volumes nearly always remain above 150 ml/kg/day.13 Inappropriate PN with steadily increasing IVF may significantly aggravate stool output. No specific physical findings can be detected apart from abdominal distension with fluid-filled intestinal and colonic loops. There are no malformations or dysmorphic features.4 Chen et al.14 recently reported prenatal molecular diagnosis of MVID for the first time. They also demonstrated a high level of amniotic fluid alpha fetoprotein (AFAFP) in the second trimester. They speculated that the AFAFP elevation might possibly be caused by in utero body fluid leakage into the amniotic fluid through fetal enteropathy.14 Kidney dysfunction may be due to long-term PN, recurrent line infections, nephrotoxic medications and dehydration.15,16
Our patient had renal tubular dysfunction likely due to dehydration initially and line infections and PN later on. The differential diagnosis included osmotic and nonosmotic diarrhea.1,4 Molecular basis of CDD due to osmotic mechanisms is clearly identified. In the nonosmotic category, only MVID, IPEX syndrome and APS-1 genes are identified so far.4 Rarely, intestinal pseudo-obstruction syndrome and Hirschsprung’s disease can present as CDD. Microscopy reveals shortened microvilli and villous atrophy with an increased number of secretory granules within enterocytes and membrane-bound inclusions on EM without crypt hyperplasia or inflammation, which was the case in our patient.5 PAS, alkaline phosphatase and immunohistochemical (pCEA and CD10) stains have proved useful.6,7 All of these four stains exhibit the same staining pattern and sensitivity, indicating that any method is useful for the diagnosis; particularly the simple and routinely used PAS stain.
In one study, morphological features together with the biomarker-highlighted apical cytoplasmic blush and inclusions were unique and diagnostic.17 They suggested that EM is not required to establish a diagnosis if these studies display typical features. As microvilli on immature crypt cells are usually normal, isolated EM of these cells should not be performed as it could lead to false negative results. In addition, the isolated finding of rudimentary or absent microvilli on enterocytes is also not sufficient to diagnose MVID.4 Our case has substantiated this with its characteristic morphohistochemical and immunohistochemical diagnostic findings and less typical findings on EM. Typical histological findings have been well described in SB biopsies, but there are few reports on their presence in the gastric mucosa.18 MVID patients are PN dependent. PN in MVID is difficult with frequent complications,8 and the risk of death or irreversible sequelae is high. SBTx is the only curative treatment.4,19
It can be performed as isolated or combined liver- SBTx, if significant liver disease exists. Early SBTx can be considered as a first choice treatment.4,19 However, the decision and timing of SBTx are difficult to arrive at because of improvements in long-term PN,20 and the complications in SBTx.8,21 Severe diarrhea, dehydration with resulting hypoperfusion can cause neurological and developmental retardation. Impaired renal function, nephrocalcinosis and growth failure are frequent.4,19 Complications of PN such as cholestasis or liver failure are very frequent. Infectious complications of the central lines are frequent etiologies of death. In one study, all 24 patients over a 14 year period were completely depended on PN.8 Thirteen children underwent SBTx (9 isolated, 4 combined with liver Tx). Patient survival rates were 63% without SBTx and 77% with SBTx. In an earlier report, 75% of 23 patients in a multicenter survey died of sepsis, liver failure, or metabolic imbalance before 9 months of age.20 Our patient exhibited severe complications and death as early as 4 months of age, which is typically described.
Conclusion
MVID is one of the most severe CDD. Severe watery diarrhea starts within the first hours or days of life resulting in severe dehydration and profound metabolic acidosis with or without renal impairment. A high index of clinical suspicion based on the background of consanguinity and use of simple and accurate diagnostic techniques (providing an alternative to EM) form the cornerstone of early diagnosis. Management includes fluid replacement and long term PN. SBTx is the only curative treatment and requires attention in long-term health planning of the country. Genetic counseling for families with an affected child is crucial. To our best knowledge, this is the first case report of using special stains in diagnosis of MVID in Oman.
Acknowledgements
The authors reported no conflict of interest and no funding was received on this work.
References
1. Canani RB, Terrin G. Recent progress in congenital diarrheal disorders. Curr Gastroenterol Rep 2011; 13:257-64.
2. Berni Canani R, Cirillo P, Terrin G. Chronic and intractable diarrhea. In: Guandalini S Ed. Essential Pediatric Gastroenterology Hepatology, and Nutrition. McGraw-Hill Mediacla Publishing Division Chicago 2005; 25-47.
3. Berni Canani R, Terrin G, Cardillo G, Tomaiuolo R, Castaldo G. Congenital diarrheal disorders: improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J Pediatr Gastroenterol Nutr 2010; 50:360-6.
4. Ruemmele FM, Schmitz J, Goulet O. Microvillous inclusion disease (microvillous atrophy). Orphanet J Rare Dis 2006; 26;1:22.
5. Bell SW, Kerner JA Jr, Sibley RK. Microvillous inclusion disease. The importance of electron microscopy for diagnosis. Am J Surg Pathol 1991; 15:1157-64.
6. Phillips AD, Szafranski M, Man LY, Wall WJ. Periodic acid-Schiff staining abnormality in microvillous atrophy: photometric and ultrastructural studies. J Pediatr Gastroenterol Nutr 2000; 30:34-42.
7. Youssef N, Ruemmele F, Goulet O, Patey N. CD10 expression in a case of microvillous inclusion disease. Ann Pathol 2004; 24:624-27.
8. Halac U, Lacaille F, Joly F, Hugot JP, Talbotec C, Colomb V, et al. Microvillous inclusion disease: how to improve the prognosis of a severe congenital enterocyte disorder. J Pediatr Gastroenterol Nutr 2011; 52:460-5.
9. Muller T, Hess MW, Schiefermeier N, Pfaller K, Ebner HL, Heinz-Erian P, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet 2008; 40:1163-5.
10. Szperl AM, Golachowska MR, Bruinenberg M, Prekeris R, Thunnissen AM, Karrenbeld A, et al. Functional characterization of mutations in the myosin Vb gene associated with microvillus inclusion disease. J Pediatr Gastroenterol Nutr 2011; 52:307-13.
11. Ruemmele FM, Müller T, Schiefermeier N, Ebner HL, Lechner S, Pfaller K, et al. Loss-of-function of MYO5B is the main cause of microvillus inclusion disease: 15 novel mutations and a CaCo-2 RNAi cell model. Hum Mutat 2010; 31:544-51.
12. Chen CP, Chiang MC, Wang TH, Hsueh C, Chang SD, Tsai FJ, et al. Microvillus inclusion disease: prenatal ultrasound findings, molecular diagnosis and genetic counseling of congenital diarrhea. Taiwan J Obstet Gynecol 2010; 49:487-94.
13. Croft NM, Howatson AG, Ling SC, Nairn L, Evans TJ, Weaver LT. Microvillous inclusion disease: an evolving condition. J Pediatr Gastroenterol Nutr 2000; 31:185-89.
14. Chen CP, Su YN, Chern SR, Wu PC, Wang W. Prenatal diagnosis of microvillus inclusion disease. Taiwan J Obstet Gynecol 2011; 50:399-400.
15. Golachowska MR, Dael CM, Keuning H, Karrenbeld A, Hoekstra D, Gijsbers CF, et al. Divergent Effects of MYO5B Mutations on Apical Brush Border and Apical Recycling Endosome Organization in Kidney and Intestinal Epithelial Cells of Microvillus Inclusion Disease Patients Presenting with Transient Renal Fanconi Syndrome. J Pediatr Gastroenterol Nutri 2011 Sep 23. [Epub ahead of print]
16. Buchman AL, Moukarzel A, Ament ME, Gornbein J, Goodson B, Carlson C, et al. Serious renal impairment is associated with long-term parenteral nutrition. J Parenter Enteral Nutr 1993; 17:438-4.
17. Al-Daraji WI, Zelger B, Zelger B, Hussein MR. Microvillous inclusion disease: a clinicopathologic study of 17 cases from the UK. Ultrastruct Pathol 2010; 34:327-32.
18. Thomas N, Pulimood AB, Kumar M, Jana AK. Microvillous inclusion disease diagnosed by gastric biopsy. Indian Pediatr 2012; 49:58-60.
19. Ruemmele FM, Jan D, Lacaille F, Cézard JP, Canioni D, Phillips AD, et al. New perspectives for children with microvillous inclusion disease: early small bowel transplantation. Transplantation 2004; 77:1024-28.
20. Colomb V, Dabbas-Tyan M, Taupin P, Talbotec C, Révillon Y, Jan D, et al. Long-term outcome of children receiving home parenteral nutrition: a 20-year single-center experience in 302 patients. J Pediatr Gastroenterol Nutr 2007; 44:347-53.
21. Lacaille F, Vass N, Sauvat F, Canioni D, Colomb V, Talbotec C, et al. Long-term outcome, growth and digestive function in children 2 to 18 years after intestinal transplantation. Gut 2008; 57:455-61.
|
|