Congenital heart disease (CHD) is the most common form of congenital anomalies1 with a reported incidence in Oman of 7.1 per 1000 births.2 Much of the literature on adults with congenital heart disease (ACHD) emerged from North America and Europe. As congenital heart surgery has been performed for more than half a century, the profile of the ACHD is largely that of operated CHD. Due to advances in the field of pediatric cardiology, cardiac surgery, and critical care, most children born with CHD survive to adulthood with the adult population with CHD exceeding the number in the pediatric population. The estimated population of ACHD in the United States is more than one million and 1.2 million in Europe.3,4 This growing population of ACHD has special medical and surgical needs due to the nature and complexity of the lesions. One of the first challenges in adults is the transition from pediatric to adult care, which should be optimized and well-structured to avoid interruption of care.5 In addition, as surgical treatment is seldom curative, this population has long-term complications including arrhythmias, infective endocarditis, stroke, pulmonary hypertension, and heart failure, which may require transplantation.6,7 Although most emerging data about ACHD was published from North America and Europe,8-10 there was a substantial increase in the number of publications related to ACHD from other parts of the world in the last few years.11-14 Several guidelines have also been published for the management of ACHD.15,16
To date, there is no published data on the spectrum of CHD in adolescents and adults in Oman. We sought to examine this for patients in Oman with CHD above the age of 13 years. In addition, we performed survival analysis for some forms of CHD among the study population.
Methods
In this retrospective cohort study data was collected from hospital records at the Royal Hospital, Muscat, which is the referral center for all CHD in Oman. All patients at or above 13 years of age who were registered in the hospital electronic health records and that had CHD were included in the study, which took place between January 2006 and January 2013. We included patients aged 13 years and above as this is the age of transition from pediatric care to adult care in Oman. The definition of CHD was based on international classification of diseases version 10.17 Patients with Marfan syndrome, hypertrophic cardiomyopathy, isolated dextrocardia with no CHD, and isolated congenital heart block with no structural abnormalities were not included in the study. If two lesions were present the more hemodynamically significant lesion was considered for primary diagnosis. The primary diagnosis was verified using echocardiography results to exclude the possibility of miscoding. The following variables were obtained in addition to the primary diagnosis: age, sex, address, date of last follow-up, and in-hospital mortality during study period. The study was approved by the research and ethics committees at the Royal Hospital, Muscat, Oman.
Statistical analysis was performed using SPSS, version 16 (Chicago, Illinois, USA) and GraphPad Prism version 5.00 (San Diego, California, USA). Categorical variables were presented as percentage and numbers, and continuous variables as mean with standard deviation (SD) or median with interquartile range (IQR). All-cause mortality during the study period was examined with the date of birth as starting time and the date of last follow up as the end-point for the analysis, using the Kaplan-Meier curve with log rank test to assess the significance. A p-value of <0.050 was considered significant.
Results
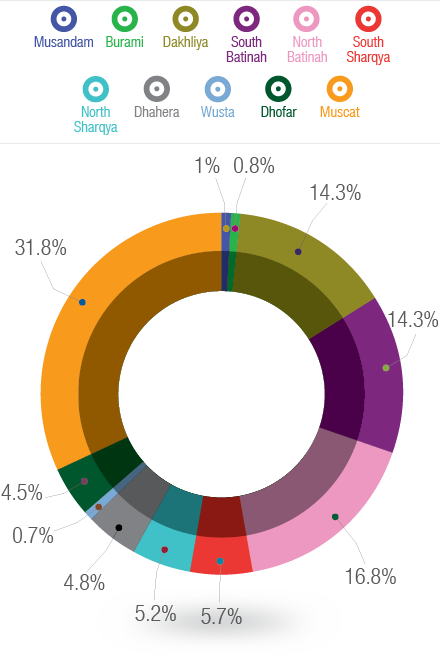
Figure 1: Geographical distribution of adolescents and adults with congenital heart diseases according to different regions in Oman.
A total of 600 patients were included in the study. Patients baseline characteristics are shown in Table 1. The mean follow up time was 4.5 years (range 2–7 years). Over half (54%) of patients were female (n=326). There were 145 (24%) patients aged 18 years or below and 455 (76%) patients aged over 18 years. The lesions specific median age for the study population is also given in Table 1. Forty patients with ventricular septal defect (32%) and 31 with atrial septal defect (12%) had unrepaired lesion at the last follow up. The distribution of all CHDs per geographical area is shown in Figure 1. The majority of patients were in capital area Muscat (32%) followed by North Batinah (17%), South Batinah (14%), and the Dakhliya region (14%). The other regions contributed to less than 6% each to the total population.
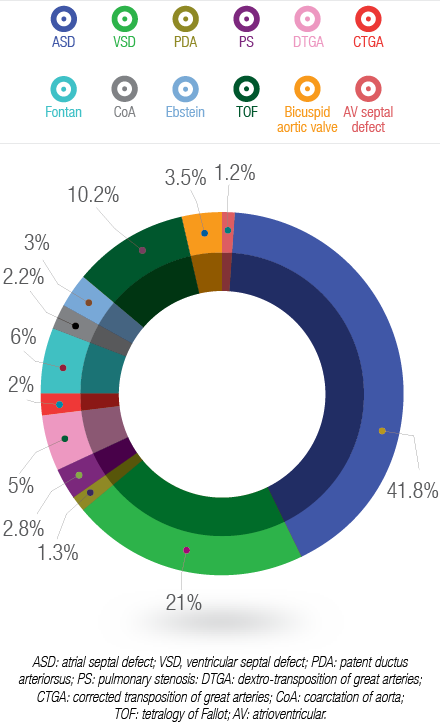
Figure 2: Spectrum of congenital heart diseases in adolescents and adults in Oman.
The spectrum of CHD per diagnosis is shown in Figure 2. The majority of patients had atrial septal defects (41.8%) followed by ventricular septal defects (21%). The following defects were more common in females than males: atrial septal defects, patent ductus arteriosus, pulmonary valve stenosis, Fontan circulation, tetralogy of Fallot, and atrioventricular septal defect. In addition, male patients had more transposition of great arteries, Ebstein’s anomaly, coarctation of aorta, and bicuspid aortic valve, and slightly more ventricular septal defects than females.

Figure 3: Kaplan-Meier curve for selected groups of congenital heart diseases.
All cause in-hospital mortality for selected CHD is shown in Figure 3. The mortality was lowest in non-cyanotic defects with patients diagnosed with atrial septal defects having the lowest mortality. The survival time among patients with ventricular septal defect was reduced compared to patients with atrial septal defects. Patients with tetralogy of Fallot and Fontan had the highest mortality with median survival time of 51 years and 31 years, respectively. This difference was statistically significant with log rank test p-value of <0.050.
Discussion
This is the first study in Oman that has described the spectrum and demographics of adolescents and adults with CHD. The available data shows that this is a young population with the majority of surviving patients having simple forms of CHD. The current overall median age for this cohort is 24 years, which is lower than what has been reported in Europe9,10 where the median age is 27 years, and 29 years in North America.16-18 We found more females with CHD in our cohort than males. This is consistent with the literature. The spectrum of CHD in this population emerging from our data is similar to other published studies that have shown that simple defects are more common in this population than cyanotic and complex defects. A previous study from Oman looking at the incidence of CHD showed no difference in the incidence of different types of CHD when compared to western countries, albeit with higher incidence of atrioventricular septal defects.2
Early mortality from CHD have been well described.18,19 It has been shown that mortality in CHD is related to either surgical mortality, or catheter related interventions in younger populations.19,20 Thus, early mortality and loss to follow-up among patients with more complex forms of CHD is a potential explanation for the predominance of simple forms of CHD in this study rather than due to lower birth incidence. To date, there is no published data from Oman on the mortality and morbidity after pediatric congenital heart surgery.
Another major finding from our study is the variability of geographical distribution of adolescents and adults with CHD in Oman; the majority of patients were in the capital area Muscat and nearby Batinah region and fewer patients living in far northern Musandam region and far south Dhofar region. Therefore, there were less registered patients in the electronic health care system from areas far from the hospital. This finding raises the issue of accessibility to health care for patients with CHD, as patients living close to the tertiary care center will tend to follow-up more than patients living far away from the center. It has been shown that the location of a CHD clinic contributed to the loss of follow-up5 with patients living close to clinic being more likely to be followed up.
An important potential explanation for this geographical variability is the presence of consanguinity in Oman. It has been shown that consanguinity, in particular, the marriage of first cousins may increase risk of CHD.20-22 However, the rate of consanguinity in Oman has not been shown to be different between different regions,22,23 thus we believe that consanguinity may not contribute to the regional variability that we have seen
in this study.
The mortality of adolescent and adults with CHD in the study is consistent with what has been reported previously with the mortality being higher in cyanotic and Fontan patients and the lowest mortality rate in patient with atrial and ventricular septal defects.9,10 However, patients with cyanotic defects and Fontan in this cohort are younger compared to published literature.23-25 The reduced survival in patients with tetralogy of Fallot and Fontan is expected knowing that patients with repaired tetralogy of Fallot and Fontan circulation are more prone to complications like arrhythmias, pulmonary hypertension and heart failure. Furthermore, we found that patients with ventricular septal defect had shorter survival time compared to patients with atrial septal defect, and this could be due to the fact that patients with unrepaired ventricular septal defects are at high risk of infective endocarditis10 and pulmonary hypertension if left untreated.26,27
As our data represent a single center experience, some patients may have presented to other regional hospitals with complications and died or lost to follow-up there. Interruption of follow-up, arrhythmias, sudden death, and infective endocarditis have been well described in this population.25,28,29 This is true for all forms of CHD including the simple forms.26-28
A major limitation of this study is the inclusion of adolescent age group to the study population; however, the pediatric age cut off in Oman is 13 years and thus these patients will be taken care in the adult health care system. The retrospective nature of this study renders it subject to recall bias. Efforts were made to minimize this by reviewing echocardiography results and making sure there is no miscoding in the diagnosis.
Table 1: Patient baseline characteristics.
|
Atrial septal defect |
251 |
32 (23–32) |
38.2/61.8 |
|
Ventricular septal defect |
126 |
21(17–21) |
25.9/24.3 |
|
Patent ductus arteriosus |
8 |
25 (18–25) |
25.0/75.0 |
|
Pulmonary valve stenosis |
17 |
28(18–28) |
17.6/82.4 |
|
D-transposition of great arteries |
30 |
21(19–21) |
66.7/33.3 |
|
L-transposition of great arteries |
12 |
24(18–24) |
58.3/41.7 |
|
Fontan circulation |
36 |
20(17–20) |
47.2/52.8 |
|
Coarctation of aorta |
13 |
21(16–21) |
69.2/30.8 |
|
Ebstein anomaly |
18 |
27(21–27) |
76.2/23.8 |
|
Tetralogy of Fallot |
61 |
21(17–21) |
45.9/54.1 |
|
Bicuspid aortic valve |
21 |
26(19–26) |
71.4/28.6 |
|
Atrioventricular septal defect |
7 |
23(20–23) |
14.3/85.7 |
*IQR: interquartile range.
Conclusion
This was the first study which looked at the spectrum of CHD outside the pediatric age group in Oman. The population emerging from this study is young compared to other developed countries with predominance of simple forms of CHD and shorter survival for those with complex forms. Further research is needed in the early outcome of pediatric heart surgery as this may impact the overall survival. In addition, research into the associated morbidities and the transition of care from pediatric to adult is needed. This can be done only by having a national registry for patients with CHD, which would help in optimizing a cost effective health care plan for
this growing population.
Disclosure
The authors declared no conflicts of interest. No funding was received for this work.
Acknowledgements
We would like to thank the medical record department at the Royal Hospital for their assistance in data retrieval and organization.
references
- van der Bom T, Zomer AC, Zwinderman AH, Meijboom FJ, Bouma BJ, Mulder BJ. The changing epidemiology of congenital heart disease. Nat Rev Cardiol 2011 Jan;8(1):50-60.
- Subramanyan R, Joy J, Venugopalan P, Sapru A, al Khusaiby SM. Incidence and spectrum of congenital heart disease in Oman. Ann Trop Paediatr 2000 Dec;20(4):337-341.
- Zomer AC, Vaartjes I, Grobbee DE, Mulder BJ. Adult congenital heart disease: new challenges. Int J Cardiol 2013 Feb;163(2):105-107.
- Greutmann M, Tobler D. Changing epidemiology and mortality in adult congenital heart disease: looking into the future. Future cardiology. 2012;8(2):171-177.
- Gurvitz M, Valente AM, Broberg C, Cook S, Stout K, Kay J, et al; Alliance for Adult Research in Congenital Cardiology (AARCC) and Adult Congenital Heart Association. Prevalence and predictors of gaps in care among adult congenital heart disease patients: HEART-ACHD (The Health, Education, and Access Research Trial). J Am Coll Cardiol 2013 May;61(21):2180-2184.
- Kovacs AH, Verstappen A. The whole adult congenital heart disease patient. Prog Cardiovasc Dis 2011 Jan-Feb;53(4):247-253.
- Mondésert B, Abadir S, Khairy P; MondŽsert B. Arrhythmias in adult congenital heart disease: the year in review. Curr Opin Cardiol 2013 May;28(3):354-359.
- Mylotte D, Pilote L, Ionescu-Ittu R, Abrahamowicz M, Khairy P, Therrien J, et al. Specialized adult congenital heart disease care: the impact of policy on mortality. Circulation 2014 May;129(18):1804-1812.
- Engelfriet P, Boersma E, Oechslin E, Tijssen J, Gatzoulis MA, Thilén U, et al. The spectrum of adult congenital heart disease in Europe: morbidity and mortality in a 5 year follow-up period. The Euro Heart Survey on adult congenital heart disease. Eur Heart J 2005 Nov;26(21):2325-2333.
- Qureshi SA, Hildick-Smith D, de Giovanni J, Clift P, Stuart G, Henderson R, et al; British Congenital Cardiac Association; British Cardiovascular Intervention Society; British Cardiovascular Society. Adult congenital heart disease interventions: recommendations from a joint working group of the British Congenital Cardiac Association, British Cardiovascular Intervention Society, and the British Cardiovascular Society. Cardiol Young 2013 Feb;23(1):68-74.
- Kempny A, Fernández-Jiménez R, Tutarel O, Dimopoulos K, Uebing A, Shiina Y, et al. Meeting the challenge: the evolving global landscape of adult congenital heart disease. Int J Cardiol 2013 Oct;168(6):5182-5189.
- Shiina Y, Toyoda T, Kawasoe Y, Tateno S, Shirai T, Wakisaka Y, et al. Prevalence of adult patients with congenital heart disease in Japan. Int J Cardiol 2011 Jan;146(1):13-16.
- Hannoush H, Tamim H, Younes H, Arnaout S, Gharzeddine W, Dakik H, et al. Patterns of congenital heart disease in unoperated adults: a 20-year experience in a developing country. Clin Cardiol 2004 Apr;27(4):236-240.
- Gnanappa GK, Ganigara M, Prabhu A, Varma SK, Murmu U, Varghese R, et al. Outcome of complex adult congenital heart surgery in the developing world. Congenit Heart Dis 2011 Jan-Feb;6(1):2-8.
- Warnes CA, Williams RG, Bashore TM, Child JS, Connolly HM, Dearani JA, et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Develop Guidelines on the Management of Adults With Congenital Heart Disease). Developed in Collaboration With the American Society of Echocardiography, Heart Rhythm Society, International Society for Adult Congenital Heart Disease, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol 2008 Dec;52(23):e143-e263.
- Marelli A, Beauchesne L, Mital S, Therrien J, Silversides CK. Canadian Cardiovascular Society 2009 Consensus Conference on the management of adults with congenital heart disease: introduction. Can J Cardiol 2010 Mar;26(3):e65-e69.
- Houyel L, Khoshnood B, Anderson RH, Lelong N, Thieulin AC, Goffinet F, et al; EPICARD Study group. Population-based evaluation of a suggested anatomic and clinical classification of congenital heart defects based on the International Paediatric and Congenital Cardiac Code. Orphanet J Rare Dis 2011;6:64.
- Marelli AJ, Mackie AS, Ionescu-Ittu R, Rahme E, Pilote L. Congenital heart disease in the general population: changing prevalence and age distribution. Circulation 2007 Jan;115(2):163-172.
- Khairy P, Ionescu-Ittu R, Mackie AS, Abrahamowicz M, Pilote L, Marelli AJ. Changing mortality in congenital heart disease. J Am Coll Cardiol 2010 Sep;56(14):1149-1157.
- Larsen SH, Emmertsen K, Johnsen SP, Pedersen J, Hjortholm K, Hjortdal VE. Survival and morbidity following congenital heart surgery in a population-based cohort of children–up to 12 years of follow-up. Congenit Heart Dis 2011 Jul-Aug;6(4):322-329.
- Becker S, Al Halees Z. First-cousin matings and congenital heart disease in Saudi Arabia. Community Genet 1999;2(2-3):69-73.
- Shieh JT, Bittles AH, Hudgins L. Consanguinity and the risk of congenital heart disease. Am J Med Genet A 2012 May;158A(5):1236-1241.
- Islam MM. The practice of consanguineous marriage in Oman: prevalence, trends and determinants. J Biosoc Sci 2012 Sep;44(5):571-594.
- Greutmann M, Tobler D, Kovacs AH, Greutmann-Yantiri M, Haile SR, Held L, et al. Increasing Mortality Burden among Adults with Complex Congenital Heart Disease. Congenit Heart Dis 2014 Jul.
- Atz AM, Zak V, Mahony L, Uzark K, Shrader P, Gallagher D, et al; Pediatric Heart Network Investigators. Survival data and predictors of functional outcome an average of 15 years after the fontan procedure: the pediatric heart network fontan cohort. Congenit Heart Dis 2015 Jan;10(1):E30-E42.
- Lin YS, Liu PH, Wu LS, Chen YM, Chang CJ, Chu PH. Major adverse cardiovascular events in adult congenital heart disease: a population-based follow-up study from Taiwan. BMC Cardiovasc Disord 2014;14:38.
- Walsh EP, Cecchin F. Arrhythmias in adult patients with congenital heart disease. Circulation 2007 Jan;115(4):534-545.
- Perry JC. Sudden cardiac death and malignant arrhythmias: the scope of the problem in adult congenital heart patients. Pediatr Cardiol 2012 Mar;33(3):484-490.
- Koyak Z, Harris L, de Groot JR, Silversides CK, Oechslin EN, Bouma BJ, et al. Sudden cardiac death in adult congenital heart disease. Circulation 2012 Oct;126(16):1944-1954.