Beta-thalassemia is an autosomal recessive inherited disorder of hemoglobin synthesis involving its β-chain. The disorder is prevalent in the Eastern Mediterranean region, including Iraq.1–3 It leads to a spectrum of clinical phenotypes ranging from β-thalassemia major (β-TM) in those homozygous or compound heterozygous for the β-thalassemia mutations to thalassemia minor in those heterozygous for it.4 The β-TM phenotype is a severe disorder associated with life-long transfusion requirements and a multitude of complications. The latter include hypothyroidism, hypoparathyroidism, hypogonadism, stunted growth, diabetes mellitus, cardiac dysfunction, and liver disease.4 The frequencies of these complications are rather variable in different β-TM cohorts and are related to adequacy of management and follow-up provided to these patients, with a much better quality of life and fewer complications in developed compared to developing countries.5
While some studies have focused on the mutation spectrum in β-TM, chelation therapy, and the health-related quality of life in Iraqi patients,5–7 there is a paucity of data on the challenges in management and complications in this developing country. We initiated this study to evaluate these complications and their management issues at a main thalassemia center in Iraq.
Methods
In this cross-sectional study, a total of 150 consecutive un-transplanted β-TM patients visiting the Duhok Thalassemia Center, Iraqi Kurdistan, for regular transfusions were recruited. This constitutes 48% of registered patients with this diagnosis at the center, after the exclusion of those who had undergone bone marrow transplantation (54 patients, 48 of whom became transfusion independent post-transplant).
In addition to a full review of the patient records, patients were interviewed at the time of enrollment and a full history and clinical exam were performed. Moreover, samples were taken for full blood counts (Swelab Alpha hematology analyzer; Boule diagnostics AB, Spanga, Sweden), and serum biochemistry (using Cobas® 6000, Roche Diagnostics, Indianapolis, IN, USA). The latter included screening for the important complications of TM. For patients >10 years old, or those with suggestive symptoms ≤ 10 years old we screened for hypothyroidism and hypoparathyroidism. For the former, serum thyroid-stimulating hormone (TSH) was assayed and, if increased, a free thyroxine (T4) test was performed. For this study, subclinical hypothyroidism was defined as increased TSH but normal free T4, while overt hypothyroidism as high TSH with reduced free T4. On the other hand, hypoparathyroidism was screened for by measuring serum calcium, if decreased, then serum parathyroid hormone was measured, and if both were reduced then the diagnosis was confirmed. Serum ferritin was measured in all patients at the time of enrollment, and the records of the serum ferritin results were reviewed. It is the policy of the center to perform hepatitis C virus (HCV), HIV 1 and 2 antibodies, and hepatitis B surface antigen (HBsAg) periodically (every six months) on all patients, and to perform HCV RNA by real-time polymerase chain reaction on all HCV antibody-positive patients, so patient records were reviewed. Patients were considered to have diabetes mellitus if they were already on insulin or oral hypoglycemic medications, otherwise, all patients > 10 years old or with suggestive symptoms had fasting blood sugar performed on two occasions, and if both were ≥ 126 mg/dL then a diagnosis of diabetes mellitus was confirmed. For the current study, male patients > 14 years, who had delayed puberty, arrest of puberty for two years, suggestive hormonal workup, or were receiving androgen therapy were considered as cases of hypogonadism. In females > 14 years old, hypogonadism was diagnosed if the patient had amenorrhea (primary or secondary). Patients were considered to have heart failure if they were already receiving anti-failure treatment, or if they satisfied the Framingham diagnostic criteria.8
The Kurdistan Board of Medical Specialties ethics committee approved the study and we obtained informed consent from all patients or their guardians.
Data were presented as mean±standard deviation (SD) if variables had a normal distribution, while median and range were used for variables with skewed distributions. The chi-square test with or without Yates correction and the non-parametric Mann-Whitney U test were used where appropriate. A two tailed p-value was used for all assessments. A p-value < 0.050 was considered significant.
Results
The median age of the enrolled patients was 13 years (range: 1–35 years), and only 2.0% were 30 years or older. The male: female ratio among patients was 1:1.2. The main characteristics of the patients are outlined in Table 1. The mean pre-transfusion hemoglobin was 8.6±1.0 g/dL, and it was maintained ≥ 9.0 g/dL in 38.7% of patients, while it was < 8.0 g/dL in 23.3%, and > 10.5 g/dL in 2.7%. In the 121 patients who were 20 years old or less, short stature (height below the third percentile) was documented in 33.9%. Thalassemic faces were noticeable in around half of the patients (50.7%). Splenectomy had been performed in 57 patients (38.0%).
Table 1: The main parameters of the Iraqi patients with β-thalassemia major, n = 150.
|
Age, years |
13 (1–35) |
|
Sex, n (males/females) |
68/82 |
|
Age at diagnosis, months |
6 (2.5–24.0) |
|
Age at first transfusion, months |
6 (3–36) |
|
Frequency of transfusions per year |
12 (8–26) |
|
Pre-transfusion hemoglobin, mean ± SD |
8.6 ± 1.0 |
|
Hepatomegaly, n (%) |
34 (22.7) |
|
Splenectomy, n (%) |
57 (38.0) |
|
Chelation therapy, n (%) |
142 (94.7) |
SD: standard deviation.
Table 2: Frequency of complications in Iraqi patients with β-thalassemia major, n = 150.
|
HCV antibody-positive |
53 (35.3) |
|
HBsAg positive |
0 (0.0) |
|
HIV antibody-positive |
0 (0.0) |
|
Hypogonadism (of 72 patients > 14 years) |
38 (52.8) |
|
Females (n = 41) |
29 (70.7) |
|
Males (n = 31) |
9 (29.0) |
|
Hypothyroidism |
11 (7.3) |
|
Hypoparathyroidism |
5 (3.3) |
|
Diabetes mellitus |
5 (3.3) |
HCV: hepatitis C virus; HBsAg: hepatitis B surface antigen.
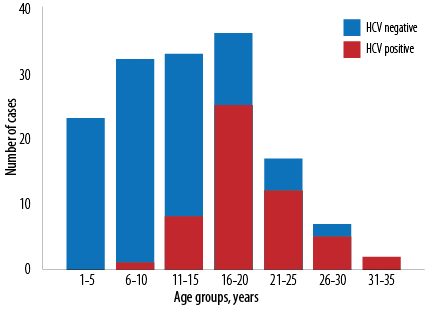
Figure 1: A histogram showing the age distribution of hepatitis C virus (HCV) seropositivity among 150 thalassemia major Iraqi patients.
The patients had a history of starting regular transfusions at a median age of six months, and they currently had a median transfusion frequency of 12 per year. HCV antibodies at the time of enrollment were detected in 53 (35.3%) of patients with HCV RNA detectable concomitantly in five. As shown in Figure 1, HCV antibody positivity increased with age (p < 0.001). Of the 53 HCV antibody-positive patients, 17 (32.1%) had not received any therapy and appear to have had spontaneous clearance and are currently HCV RNA negative. The remaining 36 patients, who had positive HCV RNA initially, received appropriate therapy with 31 (86.1%) becoming HCV RNA negative at the time of enrollment. The currently used therapy regime for HCV at the center is a combination of ledipasvir and sofosbuvir. The remaining five patients are still HCV RNA positive, and on therapy. On the other hand, HBsAg and HIV antibodies were not detectable in any of the enrollees.
The median serum ferritin was 2762 μg/L, with 53.3% of the patients having serum ferritin in excess or equal to 2500 μg/L. All patients, except eight, were on chelation therapy at the time of enrollment, including 138 (92.0%) on oral deferasirox, three (2.0%) on subcutaneous deferoxamine, and one (0.7%) on combined therapy.
Table 2 outlines the main complications encountered. The most frequent complication was hypogonadism in 52.8% of those above the age of 14 years, and it was proportionally more frequent among females compared to males (70.7% vs. 29.0%, respectively, p < 0.001). Only one patient, a female, was ever married, and none of those above 18 years had children.
As outlined in Table 2, other complications included hypothyroidism, which was encountered in 7.3% of patients all being subclinical since none had a reduced free T4. While hypoparathyroidism and diabetes mellitus were encountered in 3.3% of patients each. All diabetic patients were already on insulin therapy. Heart failure was documented in 2.7%, all of whom were receiving anti-failure therapy at the time of enrollment. No cases of arrhythmias were documented among patients at the time of enrollment.
Non-parametric tests revealed that patients with diabetes mellitus, hypoparathyroidism, and heart failure were significantly older than those without these complications [Table 3]. Although patients with hypothyroidism were older than those without, we found no statistically significant difference. Serum ferritin levels were higher in those with hypogonadism, diabetes mellitus, hypothyroidism, hypoparathyroidism, and heart failure, but none were significant, except for hypogonadism [Table 3]. Table 3 also shows that there were no significant associations between pre-transfusion hemoglobin levels and any of the complications screened for. Furthermore, it was noted that HCV infection, hypogonadism, diabetes mellitus, and heart failure were significantly more frequent in splenectomized versus non-splenectomized patients (p < 0.001, < 0.001, 0.015 and 0.039, respectively).
Table 3: Association of complications with age, serum ferritin, and pre-transfusion hemoglobin (Hb) assessed by Mann-Whitney U test.
|
Hypothyroidism |
|
0.270 |
|
0.099 |
|
0.968 |
|
Yes |
89.4 |
|
96.3 |
|
76.0 |
|
|
No |
74.4 |
|
73.9 |
|
75.5 |
|
|
Hypoparathyroidism |
|
0.009 |
|
0.182 |
|
0.285 |
|
Yes |
125.0 |
|
101.0 |
|
55.1 |
|
|
No |
73.8 |
|
74.6 |
|
76.2 |
|
|
Diabetes mellitus |
|
0.014 |
|
0.912 |
|
0.095 |
|
Yes |
122.3 |
|
77.6 |
|
107.4 |
|
|
No |
73.9 |
|
75.4 |
|
74.4 |
|
|
Heart failure |
|
0.005 |
|
0.720 |
|
0.484 |
|
Yes |
135.4 |
|
78.8 |
|
60.5 |
|
|
No |
73.9 |
|
72.4 |
|
75.9 |
|
|
Hypogonadism |
|
0.603 |
|
0.026 |
|
0.765 |
|
Yes |
35.3 |
|
41.4 |
|
37.2 |
|
|
No |
37.9 |
|
31.1 |
|
35.7 |
|
|
Hepatitis C virus |
|
< 0.001 |
|
0.043 |
|
0.196 |
|
Yes |
112.2 |
|
65.8 |
|
69.3 |
|
Discussion
β-TM constitutes an important inherited blood disorder in Iraq, with the problem being compounded by the instability and financial hardship that the country and its population has experienced over the past few decades. Our study revealed that several complications are rather common among Iraqi β-TM patients and shed some light on some of the shortcomings and challenges in the management of this disorder.
The median age of 13 years reported in the current series is not unexpected and is consistent with an earlier study from Iraq which revealed that the bulk of β-TM patients are children or adolescents.9 It is also consistent with reports from neighboring Iran, Turkey, and Egypt,10–12 though it is much lower than reports from developed countries like France, Italy, and the US.13–15 This is consequent to the higher quality of care and follow-up in the latter countries as opposed to our region. One important parameter that is reflective of the latter is the lower mean pre-transfusion hemoglobin (8.6 g/dL) in our series compared to most other studies from developed countries like Italy and the US,14–16 where pre-transfusion hemoglobins were maintained between 9–10.5 g/dL as per thalassemia international federation guidelines.17 In our series, only 38.7% of the patients had pre-transfusion hemoglobin ≥ 9.0 g/dL compared to 89% in the US.14 Such less than adequate transfusion therapy coupled by lax adherence to timely transfusions by patients/families is reflected by the prominence of skeletal changes (thalassemic faces) noted in more than half of our patients, and the higher than expected frequency of short stature, which was also seen in Egyptian patients.12 The need to review the locally adopted management guidelines at the thalassemia center and promote adherence to thalassemia international federation most recent guidelines is paramount.17 Furthermore, steps to better educate patients and their guardians on the need for regular and timely transfusion should be undertaken.
Among the transfusion-related infections in β-TM in our series, HCV stands out at 35.3%, which is similar to rates reported from the US (35%), but higher than rates reported from France, Turkey, Lebanon, Egypt, and Iran (4.1–26%),12–14,18–20 and lower than rates reported from Pakistan (49%).21 An earlier study from Southern Iraq reported higher rates of HCV seropositivity at 42.5% among transfusion-dependent thalassemia patients, though they noted that the rates are declining progressively.22 Such a high frequency of HCV among our enrollees underscores the need to introduce more sensitive screening for HCV at the blood bank, including the use of nucleic acid amplification technology to reduce the rate of transmission.23 It is important to note that the rate of spontaneous clearance of HCV in the current cohort of 32.1% is similar to that reported in North America at 33%, both these results are higher than the rates reported worldwide at 10–20%.14,24 On the other hand, the absence of HBsAg and HIV antibody-positive cases in our study is a testament to the sensitivity of pre-transfusion testing and the mandatory vaccination policy implemented for the last three decades in the country for HBV, and very low prevalence of HIV infection in the population, respectively.25 The respective figures for HBsAg and HIV in an earlier study on TM Iraqi patients were 0.4% and 0%.9 Prevalence rates of HBsAg in other TM cohorts ranged from 0.2% to 2.5% in Iranian, French, and North American TM patients, while those for HIV ranged between 0.2% and 2.0% in Iran, Italy, France, and North America.13,14,16,18
The high rate of use of oral chelation therapy (deferasirox) among our patients is mainly due to its ease of use, and its provision free of charge by the Ministry of Health to all thalassemia centers in the country. A recent study from our center showed a significant though modest reduction in serum ferritin levels after a median follow-up of two years, and compliance of nearly 77% and suggested the need to optimize the dose of this medication better.26 Previous studies have confirmed the value of deferasirox in reducing serum ferritin and liver iron content.27 The median ferritin in our patients is nearer to figures reported in the UAE and Turkey but is higher than those reported in France and North America and lower than those reported in some Iranian and Egyptian studies.10–14,28 To a great extent, this reflects the adequacy of chelation therapy in developed countries compared to some developing countries and is probably also related to the availability and type of chelation administered and literacy of patients or their guardians in different countries. The need to introduce more robust tests for evaluating iron overload, including liver iron content testing and T2* magnetic resonance imaging (MRI) is important. The former, being a rather invasive procedure, is only provided currently for those contemplating bone marrow transplants at our center while the non-invasive T2*MRI to evaluate cardiac iron has not yet become available as an option to our physicians. The latter should be made a priority given the large number of registered patients with significant iron overload, as serum ferritin may not accurately reflect the extent of iron overload, particularly cardiac overload.29
Among the well-documented complications of β-TM related to iron overload is hypothyroidism, which varies in frequency in different studies worldwide between 4.25% and 10.8%, so our rate of 7.3% is not unexpected.10–14,16,18,28,30,31 On the other hand, the rate of hypoparathyoidism at 3.3% is less than the rates reported in some other populations of 4.0–10.5%.10,12,14,18,28,30,31 Diabetes mellitus is another recognized complication and varies in frequency between 2.1% and 26.7%, so our rate of 3.3% is near the lower end of the range.10–14,16,18,28,30,31 Hypogonadism is the most frequently reported complication due to iron overload in almost all thalassemic populations in developed and underdeveloped countries and ranges from 28.7% to 73.3%, so our figure of 52.8% is expected.10,13,14, 16,18,28,30–32 Low frequencies of parenthood have been reported among thalassemia patients in many studies.13,14 The main reason is likely to be a delay in initiating hormonal therapy to induce puberty, in addition to the social and physical impact of the disease, which leads to less likelihood of marriage.
Heart failure has been reported in a variable proportion of patients, and we would expect it to increase with age.14 The reported rates in β-TM ranged from 1.8% in the UAE (for heart failure and arrhythmias),28 6.8% in Italy,16 9.7% in France,13 to 10% in the US (for cardiac disease requiring therapy).14 Our rate is nearer to the UAE figure, most likely due to the younger cohort in these two populations. Other cardiac complications like pulmonary hypertension were investigated in the same center by another group of researchers simultaneously and revealed a rate of 9.0% in β-TM patients older than eight years (Dr. Ameen M. Mohammad, personal communication).
It should be noted that while we documented significant association of most complications with older age, we failed to find such an association with serum ferritin. Such an observation is shared by earlier French, Omani, and Turkish β-TM studies.11,13,31 This observation may be related to the limitation imposed by the number of those with the respective complications, and the relative inadequacy of serum ferritin as a measure of iron overload. One notable exception was the association of hypogonadism with higher serum ferritin, an observation shared by an earlier Iranian study.30 Another interesting finding is the association of splenectomy with various complications in β-TM, which has also been reported by earlier studies and may be related to a multitude of possible causes, including more exposure of various organs to free iron radicals in the splenectomized compared to the non-splenectomized patients.11,33–36
Bone marrow transplantation is the only curative treatment for β-TM, and for the last decade or so, the local government has offered financial support to β-TM patients with compatible donors to do the procedure in neighboring countries, leading to increasing numbers of patients taking this option. Currently, 14.8% of our β-TM population have been transplanted (with 88.9% becoming transfusion independent), and it is hoped that with the establishment of a transplantation center in the region, more patients would embark on this option.
Limitations of the current study include that it is a cross-sectional and not a longitudinal one, and partically based on reviewing the records of enrolled patients, which has obvious drawbacks. Another limitation was the unavailability of T2*MRI, which limited the ability to accurately determine the iron overload status of the patients. Furthermore, while the study focused on the main complications encountered in β-TM, some complications like pulmonary hypertension and osteoporosis were not focused upon since fellow researchers at the center were studying them, and their results will complement our observations as they get published.
Conclusions
Patients with β-TM in Iraq have a lower median age, and are less adequately transfused with more prominent consequent skeletal changes and growth retardation. The recognized complications of β-TM occur at comparable frequencies to other β-TM populations. The need to ensure a more adequate and timely transfusion and chelation are paramount, and the need to introduce more sensitive methods for donor blood HCV screening and more robust techniques to assess iron overload should be emphasized to improve overall survival and quality of life.
Disclosure
The authors declared no conflicts of interest. No funding was received for this study.
references
- 1. Yahya HI, Khalel KJ, Al-Allawi NA, Helmi F. Thalassemia genes in Baghdad, Iraq. East Mediterr Health J 1996;2(2):315-319.
- 2. Al-Allawi NA, Al-Dousky AA. Frequency of haemoglobinopathies at premarital health screening in Dohuk, Iraq: implications for a regional prevention programme. East Mediterr Health J 2010 Apr;16(4):381-385.
- 3. Hamamy HA, Al-Allawi NA. Epidemiological profile of common haemoglobinopathies in Arab countries. J Community Genet 2013 Apr;4(2):147-167.
- 4. Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis 2010 May;5(1):11.
- 5. Mikael NA, Al-Allawi NA. Factors affecting quality of life in children and adolescents with thalassemia in Iraqi Kurdistan. Saudi Med J 2018 Aug;39(8):799-807.
- 6. Al-Allawi NA, Hassan KM, Sheikha AK, Nerweiy FF, Dawood RS, Jubrael J. Thalassemia mutations among transfusion-dependent thalassemia Mmajor patients in Northern Iraq. Molecular Biology International 2010;2010:479282.
- 7. Abdul-Zahra HA, Hassan MK, Ahmed BA. Health-related quality of life in children and adolescents with b-thalassemia major on different iron chelators in Basra, Iraq. J Pediatr Hematol Oncol. 2016 Oct;38(7):503-511.
- 8. Ho KK, Pinsky JL, Kannel WB, Levy D. The epidemiology of heart failure: the Framingham study. J Am Coll Cardiol 1993 Oct;22(4)(Suppl A):6A-13A.
- 9. Kadhim KA, Baldawi KH, Lami FH. Prevalence, incidence, trend, and complications of thalassemia in Iraq. Hemoglobin 2017 May;41(3):164-168.
- 10. Yaghobi M, Miri-Moghaddam E, Majid N, Bazi A, Navidian A, Kalkali A. Complications of transfusion-dependent b-thalassemia patients in Sistan and Baluchistan, South-East of Iran. Int J Hematol Oncol Stem Cell Res 2017 Oct;11(4):268-272.
- 11. Isik P, Yarali N, Tavil B, Demirel F, Karacam GB, Sac RU, et al. Endocrinopathies in Turkish children with Beta thalassemia major: results from a single center study. Pediatr Hematol Oncol 2014 Oct;31(7):607-615.
- 12. Hassan T, Zakaria M, Fathy M, Arafa M, El Gebaly S, Emam A, et al. Association between genotype and disease complications in Egyptian patients with beta thalassemia: a cross-sectional study. Sci Rep 2018 Dec;8(1):17730.
- 13. Thuret I, Pondarré C, Loundou A, Steschenko D, Girot R, Bachir D, et al. Complications and treatment of patients with b-thalassemia in France: results of the national registry. Haematologica 2010 May;95(5):724-729.
- 14. Cunningham MJ, Macklin EA, Neufeld EJ, Cohen AR; Thalassemia Clinical Research Network. Complications of beta-thalassemia major in North America. Blood 2004 Jul;104(1):34-39.
- 15. Chirico V, Lacquaniti A, Salpietro V, Luca N, Ferraù V, Piraino B, et al. Thyroid dysfunction in thalassaemic patients: ferritin as a prognostic marker and combined iron chelators as an ideal therapy. Eur J Endocrinol 2013 Oct;169(6):785-793.
- 16. Borgna-Pignatti C, Rugolotto S, De Stefano P, Zhao H, Cappellini MD, Del Vecchio GC, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 2004 Oct;89(10):1187-1193.
- 17. Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V. Guidelines for the management of transfusion dependent thalassemia (TDT). 3rd ed. Thalassemia International Federation, Nicosia; 2014. p. 30.
- 18. Hajipour M, Soltani M, Najafifar R, Rezaeian S, Bistoon-beigloo S, Valadbeigi T, et al. Complications of beta-thalassemia patients and their related factors in Iran. International Journal of Development Research 2015;5(7):5009-5014.
- 19. Ramia S, Koussa S, Taher A, Haraki S, Klayme S, Sarkis D, et al. Hepatitis-C-virus genotypes and hepatitis-G-virus infection in Lebanese thalassaemics. Ann Trop Med Parasitol 2002 Mar;96(2):197-202.
- 20. Jaiswal SP, Chitnis DS, Jain AK, Inamdar S, Porwal A, Jain SC. Prevalence of hepatitis viruses among multi-transfused homogenous thalassaemia patients. Hepatol Res 2001 Mar;19(3):247-253.
- 21. Din G, Malik S, Ali I, Ahmed S, Dasti JI. Prevalence of hepatitis C virus infection among thalassemia patients: a perspective from a multi-ethnic population of Pakistan. Asian Pac J Trop Med 2014;7S1:S127-S133.
- 22. Najim OA, Hassan MK. Prevalence of hepatitis C virus seropositivity among multitransfused patients with hereditary anemias in Basra, Iraq. Iraqi J Hematology 2018;7(1):39-44.
- 23. Engelfriet CP, Reesink HW. International forum: implementation of donor screening for infectious agents transmitted by blood by nucleic acid technology. Vox Sang 2002;82(2):87-111.
- 24. Mazzeo C, Azzaroli F, Giovanelli S, Dormi A, Festi D, Colecchia A, et al. Ten year incidence of HCV infection in northern Italy and frequency of spontaneous viral clearance. Gut 2003 Jul;52(7):1030-1034.
- 25. Massah O, Moradi A, Farhoudian A, Amini-Lari M, Joulaei H, Daneshmand R. HIV programs in Iran (Persia), Iraq and Saudi Arabia: a brief review of current evidence in West and Southwest Asia. Addict Health 2016 Jul;8(3):136-144.
- 26. Hassan LJ. Deferasirox effect on serum ferritin in patients with chronic hemolytic anemia. Pediatric hematology diploma dissertation. College of Medicine, University of Duhok, Iraq; 2018.
- 27. Taher A, Elalfy MS, Al Zir K, Daar S, Al Jefri A, Habr D, et al. Importance of optimal dosing ≥ 30 mg/kg/d during deferasirox treatment: 2.7-yr follow-up from the ESCALATOR study in patients with b-thalassaemia. Eur J Haematol 2011 Oct;87(4):355-365.
- 28. Belhoul KM, Bakir ML, Kadhim AM, Dewedar HE, Eldin MS, Alkhaja FA. Prevalence of iron overload complications among patients with b-thalassemia major treated at Dubai Thalassemia Centre. Ann Saudi Med 2013 Jan-Feb;33(1):18-21.
- 29. Soltanpour MS, Davari K. Correlation of cardiac and hepatic hemosiderosis as measured by T2* MRI technique with ferritin levels and hemochromatosis gene mutations in Iranian patients with beta thalassemia major. Oman Med J 2018 Jan;33(1):48-54.
- 30. Shamshirsaz AA, Bekheirnia MR, Kamgar M, Pourzahedgilani N, Bouzari N, Habibzadeh M, et al. Metabolic and endocrinologic complications in beta-thalassemia major: a multicenter study in Tehran. BMC Endocr Disord 2003 Aug;3(1):4.
- 31. Mula-Abed WA, Al Hashmi H, Al Muslahi M, Al Muslahi H, Al Lamki M. Prevalence of endocrinopathies in patients with Beta-thalassaemia major - a cross-sectional study in Oman. Oman Med J 2008 Oct;23(4):257-262.
- 32. Derchi G, Formisano F, Balocco M, Galanello R, Bina P, Dessì C, et al. Clinical management of cardiovascular complications in patients with thalassaemia major: a large observational multicenter study. Eur J Echocardiogr 2011 Mar;12(3):242-246.
- 33. Meloni A, Pistoia L, Ciancio A, Santamaria V, Sorrentino F, Cirotto C, et al. Splenectomy is a risk factor for cardiac complications in TM. Blood 2017;130(Supp 1):2225.
- 34. Chuncharunee S, Teawtrakul N, Siritanaratkul N, Chueamuangphan N. Review of disease-related complications and management in adult patients with thalassemia: a multi-center study in Thailand. PLoS One 2019 Mar;14(3):e0214148.
- 35. Skordis N, Michaelidou M, Savva SC, Ioannou Y, Rousounides A, Kleanthous M, et al. The impact of genotype on endocrine complications in thalassaemia major. Eur J Haematol 2006 Aug;77(2):150-156.
- 36. Alavian SM, Tabatabaei SV, Lankarani KB. Epidemiology of HCV infection among thalassemia patients in Eastern Mediterranean countries: a quantitative review of literature. Iran Red Crescent Med J 2010;12(4):365-376.