Cystic fibrosis (CF) is a multisystem disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene.1 It is the most common life-limiting, autosomal recessive disorder among white individuals, with an average incidence of 1:3000 live births.2 CFTR protein is expressed on the apical epithelium of the intestines, pancreas, and biliary duct system where it regulates chloride and bicarbonate secretion and therefore regulating the fluidity and alkalinity of the bile.1,3 Homozygosity of the mutant CFTR gene results in an alteration in bile secretion (its hydration and alkalinity) resulting in viscous and acidic secretions.1,4,5 This leads to partial or complete obstruction in various hollow epithelial-lined structures of the gastrointestinal (GI) tract and is responsible for most GI, pancreatic, and hepatobiliary manifestations of CF.4,5
Advances in specialized patient care have led to increasing the life expectancy of patients with CF.6 An estimated median survival of more than 50 years in patients born in the UK in 2000 and after is becoming more likely.7 With increased life expectancy, CF-related liver disease has increasingly been reported.8 Cystic fibrosis liver disease (CFLD) is a chronic inflammatory sclerosing cholangiopathy commonly presenting before the age of 20 years and may evolve into advanced liver disease, biliary cirrhosis, portal hypertension, and liver decompensation.1,6 Thus, early identification of liver involvement and prompt therapeutic intervention is becoming a pertinent clinical issue.8
Epidemiology of CFLD
Since the definition of CFLD is not universally unified, the true prevalence of CFLD is difficult to determine.9 However, CFLD is estimated to occur in around 10% of patients with CF and is regarded as the third leading cause of death in this population after respiratory and transplantation complications.1,10,11
Nonspecific increase in transaminases has been documented in more than 50% of infants, which often resolves by 2 to 3 years of age, and does not seem to reappear later in life or affect the future development of CFLD.12 However, patients with CF who develop significant liver disease and cirrhosis may present with normal or only slightly elevated liver enzymes.8,12 The incidence of CFLD has been estimated at 2.5/100 patient-years during the first 10 years of life, with the possibility of sharp decline during the second decade.13 Long-term follow-up studies have found the prevalence of abnormal hepatic histopathology in patients with CF ranges from 27% to 41%.12,13 Focal biliary cirrhosis is considered the most common CFLD, with a frequency of 20% to 30%.8,14 Approximately 5% to 10% of children with CF develop cirrhosis before or during puberty.12,13 Most of these patients eventually progress to portal hypertension and present with complications related to CFLD during the second decade of life.15,16 CFLD accounts for 2.5% of overall CF mortality causes.10
Few CF prevalence studies are available from Arab countries. These suggest that CF is undiagnosed in these populations.17 Frequencies of 1:2664 in Egypt,17 1:2650 in Jordan,18 1:2560 in Kuwait,18 1:5800 in Bahrain,19 and 1:15 876 in the UAE20 have been reported. In Saudi Arabia, a report of 36 patients with CF between 1986 and 1992 revealed a 25% prevalence of liver disease in patients with CF.21 Reports indicate the possible presence of higher incidence rate in Arab population particularly given the high consanguinity rate in the range of 25–60%.22
Pathogenesis of CFLD
CFTR is expressed on apical membranes of cholangiocytes and gallbladder epithelial cells and plays a critical role in chloride and water secretion to support normal bile formation and flow.1,4,5 Homozygosity of the mutant CFTR gene results in viscous and acidic secretions secondary to deficient surface fluid and bicarbonate efflux.1,4,5,23 Viscous and inspissated bile in CF causes ductular obstruction and hepatotoxicity from retained bile components, leading to inflammation and subsequent fibrosis and cirrhosis.1,4,5 In the majority of patients, the disease leads to focal changes with no clinical significance.23 However, multilobular cirrhosis develops in a small subset of patients leading to the development of portal hypertension, splenomegaly, and hypersplenism.23 The risk of developing multilobular cirrhosis is altered by non-CFTR genetic and environmental modifiers.23 These modifiers include factors such as history of meconium ileus, male sex, severe class I-III mutations genotype, histocompatibility complex antigens, infective agents, protease inhibitor gene, nutritional state, antioxidants deficiency, viral hepatitis, and certain therapeutic methods.13,24,25 However, the role of such factors in the development of CFLD is controversial.26 In addition, correlation to the degree of pancreatic sufficiency has been proposed where CFLD is generally not observed in patients with pancreatic sufficiency.27
The phenotypic variability of CFLD in patients with CF may be explained by other insults and factors.28 Liver inflammation in predisposed individuals can be caused by the initiation of intrinsic immune responses to enteric microflora endotoxins. In the liver, gut-derived endotoxins are normally cleared by the hepatocytes into the bile. In normal situations, the biliary epithelium expresses
endotoxin tolerance that prevents an excessive inflammatory response.28 In CF mice models, increased intestinal permeability and release of bacterial endotoxins, triggers biliary damage and inflammation.29 In these mice models, using oral antibiotics to produce bowel decontamination diminished hepatic inflammation.29 Furthermore, cholangiokcytes in CF mice express higher nuclear factor-kB (NF-kB) activity and NF-kB-dependent inflammatory cytokines when exposed to microflora endotoxins.1 It is illustrated that CFTR controls inflammation in secretory epithelia. Consequently, CFTR deficiency in cholangiocytes modifies the normal response to gut endotoxins in the epithelium with a stronger inflammatory reaction thus contributing to the pathogenesis of CFLD.1,11
Non-cirrhotic portal hypertension (NCPH) is increasingly recognized in a subset of patients with CF as more patients have a longer lifespan. It is thought to be due to obliterative venopathy with dense fibrosis within portal vein branches.11
Clinical manifestations of CFLD
CFLD manifestations vary ranging from mild asymptomatic hypertransaminasemia to liver cirrhosis [Table 1].8 The most common presentation of CFLD is an incidental finding of hepatomegaly with or without splenomegaly, jaundice, biliary colic, and abnormal liver function tests.8 Other manifestations of CFLD may include micro-gallbladder, cholelithiasis, and sclerosing cholangitis.8,25 Recent studies show reports of NCPH in patients with CF.11
In infants with CF, liver involvement may manifest in the form of cholestasis. In about 50% of cases, it co-occurs with meconium ileus and parenteral nutrition.24 Cholestasis usually resolves within the first few months of life spontaneously. However, in some cases, liver fibrosis may develop.24 In older children, CFLD may manifest as hepatic steatosis with or without hepatitis, focal biliary cirrhosis, and multilobular cirrhosis.24 In adolescence, apparent disease may develop around puberty and can progress to end-stage liver disease.8
The clinical manifestations of advanced CFLD include jaundice, coagulopathy, ascites, and the development of portal hypertension.27 Biochemical indicators of liver failure such as increased level of bilirubin, lower albumin level, and prolonged prothrombin time appear late in advanced CFLD.27
Table 1: Clinical manifestations of cystic fibrosis liver disease.8
|
Asymptomatic elevation of liver enzymes |
Common |
|
Hepatic steatosis |
25–60 |
|
Focal biliary cirrhosis |
20–30 |
|
Multilobular biliary cirrhosis |
10 |
|
Neonatal cholestasis |
< 10 |
|
Cholelithiasis and cholecystitis |
15 |
|
Micro-gallbladder |
30 |
|
Portal hypertension |
2–5 |
Diagnosis of CFLD
Early diagnosis of CFLD is important as symptoms may appear late with advanced hepatobiliary system involvement.27 The diagnosis of CFLD is suspected in patients with CF and manifestations of liver involvement. In such cases, CFLD is diagnosed after excluding other etiologies of liver disease [Table 2].8 Laboratory markers of CFLD manifesting as cholestasis and abnormal levels of transaminases are commonly observed. However, they are not sensitive or specific for CFLD and may correlate poorly with the degree of severity of liver involvement.27 Diagnosis of early signs of liver failure can be difficult as hepatic function may be retained until late phases. Prothrombin time and coagulation factors should be measured at least once a year to detect early signs of liver failure.8 Reduction in coagulation factor V is often noted in patients with splenomegaly, possibly as a result of splenic consumption.8
Noninvasive fibrosis biomarkers are novel and inexpensive serologic tests that provide an approach to investigate fibrosis in CFLD in its early stages. Examples of these biomarkers are the aspartate transaminase (AST)-to-platelet ratio index (APRI) and the fibrosis-4 index (FIB-4).11 APRI appears to be superior to FIB-4 in the assessment of fibrosis, cirrhosis, and portal hypertension.11
Table 2: Causes of acute or chronic liver disease in cystic fibrosis patients showing hepatic abnormalities.8
|
Acute/chronic viral hepatitis |
Serology for HAV, HBV, HCV, EBV, CMV, adenovirus, HHV 6, parvovirus |
|
α1 antitrypsin deficiency |
Serum α1 antitrypsin level, including phenotype |
|
Autoimmune hepatitis |
Non-organ specific autoantibodies (SMA, anti-LKM1, LC1) |
|
Celiac disease |
Total IgA, IgA anti-tissue transglutaminase |
|
Wilson disease |
Ceruloplasmin, serum copper, 24 hour urinary copper |
|
Genetic hemochromatosis |
Iron, ferritin, transferrin binding capacity |
Increasing evidence implicates dysregulated microRNA (miRNA) expression in a range of human pathologies, including liver disease and CFLD.30–35 Studies investigated the use of miRNAs for the detection of CFLD in children when compared to children with CF without liver disease and controls.34 Therefore, miR-122 may be of use for the diagnosis of CFLD in children.11,34,35
Ultrasound (US) takes a pivotal role in the diagnosis of CFLD, where it can assess liver parenchyma changes such as fibrosis, cirrhosis, steatosis, biliary duct abnormalities, and abnormalities of blood flow in the portal vein.27 Abnormal echogenicity found in US precedes clinical and biochemical manifestations of CFLD36,37 suggesting that routine US may be a valuable tool for detection of early CFLD.8
CT and MRI are useful tools to diagnose fibrosis, steatosis, biliary abnormalities, and focal lesions.8 Additionally, magnetic resonance cholangiopancreatography (MRCP) can be utilized for detection of early intrahepatic biliary duct abnormalities in patients with CF.8,38
Transient elastography (TE) or FibroScan is a noninvasive measure of liver stiffness that has been validated for the diagnosis of liver disease in viral hepatitis, alcoholic liver disease, and nonalcoholic fatty liver.8,11 Recent reports have illustrated the usefulness of TE in the diagnosis and surveillance of fibrosis in CFLD.39,40
Liver biopsy is the gold standard for the diagnosis of CFLD, its type of involvement, severity, and staging.8,11 Nonetheless, a liver biopsy may underestimate the disease severity because of the patchiness of lesions.8,11 Steatosis is reported to be the most common liver histopathologic finding in CFLD (25–60%), followed by focal biliary cirrhosis (30%) [Table 1].8,12
Table 3: Diagnostic criteria of cystic fibrosis liver disease (CFLD).8,41
|
Diagnosis of CFLD should be considered if one of the following categories is present: |
Not included |
Liver biopsy indicating pathology
or radiologic evidence of diffuse liver disease or cirrhosis |
|
Diagnosis of CFLD should be considered if two or more of the following categories are present: |
|
|
Physical examination |
X |
Not included |
|
Blood tests |
X |
X |
|
Imaging |
X |
X |
|
Transient elastography |
Not included |
X |
|
Histology |
X |
As above |
Diagnostic criteria of CFLD
Diagnostic criteria for CFLD have been proposed by Debray et al8 and Koh et al41 [Table 3]. In such criteria, the diagnosis of CFLD depends on multiple parameters, such as physical examination, liver function tests, imaging, noninvasive biomarkers, TE, and liver biopsy among others [Table 3].
Debray’s criteria suggest that the presence of CFLD should be considered if at least two of the following four elements are present: abnormal physical examination manifested as hepatomegaly and/or splenomegaly confirmed by US, persistent elevations of liver function tests (defined as abnormal values measured at least three successive times over a period of 12 months and after excluding other causes of liver disease), US confirmation of liver manifestation or signs of portal hypertension or biliary abnormalities, and abnormal histology on liver biopsy.8
On the other hand, Koh’s new criteria are based on the number of elements present at a time of evaluation.41 According to Koh’s new criteria, the diagnosis of CFLD should be considered if only one element such as liver biopsy indicating pathology or if radiologic evidence of diffuse liver disease or cirrhosis is present.41 In addition, and based on the growing evidence of TE and noninvasive biomarkers utilization in CFLD, Koh proposed adding such criteria to facilitate the diagnosis of CFLD. Based on that, Koh’s new criteria propose that the diagnosis of CFLD should be considered if two or more of the following elements are present: at least two persistently abnormal AST, alanine transaminase (ALT), alkaline phosphatase (ALP), or gamma glutamyl transpeptidase (GGT) (defined as abnormal levels on multiple dates over minimum of two consecutive years), evidence of hepatomegaly, splenomegaly, or portal hypertension by imaging, abnormal TE at any time, persistently abnormal APRI, FIB-4, or AST to ALT ratio (defined as abnormal levels on multiple dates over minimum of two consecutive years). However, validation of such criteria is awaited.41
Management of CFLD
CF patients with CFLD require annual follow-up by gastroenterologists or hepatologists to evaluate the progression of CFLD [Figure 1].8 The follow-up would necessitate screening for the development of portal hypertension and other complications via annual physical examinations, liver function tests, and abdominal imaging.8,11 There are no existing evidence-based guidelines or specific recommendations for the prevention or management of CFLD.1,11,42 Therefore, once CFLD is diagnosed, therapy is directed towards lowering the impact of complications.1,11,42
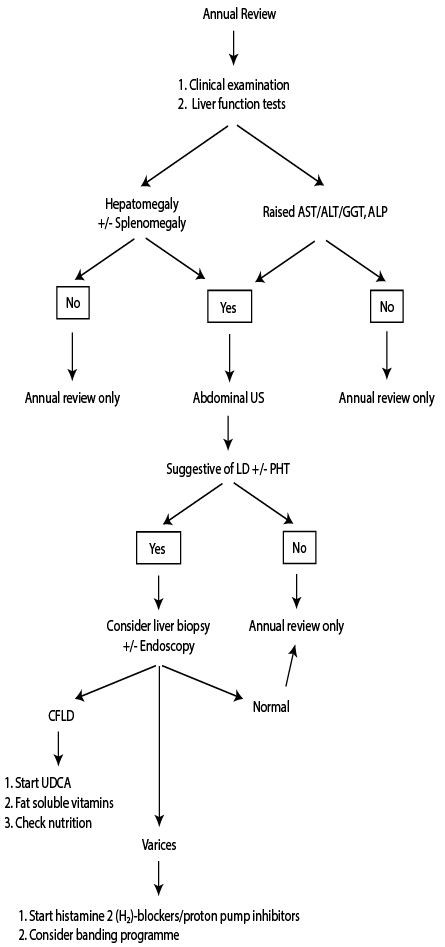
Figure 1: Flow chart for the investigation and management of cystic fibrosis-associated liver disease.8
Management of CFLD emphasizes on optimizing nutritional status via adequate caloric intake, fat-soluble vitamin supplementation, pancreatic enzyme replacement therapy, and avoiding hepatotoxic medications.42 Ursodeoxycholic acid (UDCA) is the only available treatment that may prevent the progression of CFLD.10 The physiological benefits of UDCA comprise improving bile flow and non-CFTR biliary chloride channels and protecting hepatocytes from the toxicity of accumulating bile acids.43 Reports have documented improvement in hepatic excretory function and liver histology in patients with CFLD after starting UDCA.44,45 UDCA treatment aims to delay liver disease progression. Therefore, treatment should be started as soon as the diagnosis of CFLD is made or in patients who are at risk such as patients with history of meconium ileus.8 UDCA might be of use in preventing sludge and stone formation, but it has no role in the management of gallstones.45 When UDCA is initiated, testing of liver enzymes serum levels and cholestasis markers should be performed three and six months from initiation of UDCA to evaluate its efficacy. However, the benefit of routine use of UDCA in CF is uncertain due to the scarcity of long-term studies.1,44 In addition, not all CFLD is caused by cholestasis.11
A vital aspect of CFLD management is nutritional support. The development of liver disease may worsen CF associated malnutrition by aggravating fat and protein malabsorption.23 Nutritional therapy involves improving energy intake to 150% of estimated requirement.23 In addition, protein supplementation is required in patients without signs of liver failure. Avoiding salt supplementation in advanced CFLD is essential to avoid precipitating development of ascites.23 Adequate pancreatic enzymes must be provided allowing for optimal absorption of fat and protein.23 Patients with CF and exocrine pancreatic insufficiency require fat-soluble vitamin supplements of vitamin A, vitamin E, and vitamin D, and occasionally vitamin K is required. In patients with anorexia, enteral tube feeding may be necessary to ensure adequate caloric and supplementation intake.23 Because of the risk of bleeding, gastrostomy feeding is not recommended in patients with advanced CFLD, varices, or portal gastropathy.8
Patients with cirrhosis or signs of portal hypertension should be screened for esophageal varices via upper GI endoscopy at diagnosis and every two to three years if initial screening is normal. Signs suggestive of GI bleeding should necessitate an upper GI endoscopy. Portal hypertension and variceal bleeding require management of acute episode and long-term therapy protocols.16 Transjugular portosystemic and surgical portosystemic shunts are indicated in refractory cases or as a bridge to liver transplantation.16
Patients with CF cirrhosis will require annual US of the liver and serum alpha-fetoprotein level measurement to screen for the possible development of hepatocellular carcinoma.46,47
Liver transplantation (LT) should be considered in CF patients with decompensated liver disease, cirrhosis and portal hypertension, progressive deterioration of pulmonary function, and severe malnutrition that is refractory to nutritional support.8,16,48–50 Isolated LT in CFLD has been reported to help improve the respiratory function and general patient condition.50,51 Careful patient assessment for either isolated LT or in combination of lung transplantation should be practiced.50 The five-year survival rates for children and adults undergoing LT were reported to be 85.8% and 72.7%, respectively.50,51 Although the survival rate is satisfactory, it is lower than that of LT for other etiologies, which could be explained by the lower nutritional status, decline in pulmonary function, and infection in this patient population.51 Overall, there is a survival advantage in LT compared to patients remaining on the waiting list.51 However, this advantage may not be seen in patients with isolated variceal bleeding as manifestation of CFLD.16 Mortality is increased in children who are below the fifth percentile for height and weight at time of LT.51 Progressive pulmonary disease, poor nutritional status and lower body mass index, and infections have been reported to contribute to LT late mortality.51
Novel therapies
Small molecules that increase either the number or the opening time of CFTR channels on the cell surface have now been identified and successfully trialed in patients with CF.52,53 Ivacaftor (VX770) is a CFTR potentiator administered to patients with a 551G>D mutation that affects channel gating by improving the opening time of the mutant channel, and hence increasing its ion transport. It has shown effect through the improvement in sweat chloride concentration, lung function, body mass index, and decreased exacerbation rate.1,54 Ivacaftor therapy also was noticed to induced change in the GI pH profile,2 that led to a reduction in the daily pancreatic enzyme supplements dose.54 Lumacaftor (VX-809) has been approved, in combination with ivacaftor, for patients homozygous for F508D.28 It acts as a CFTR channel corrector that suppresses misfolding of the CFTR protein, allows the CFTR to bypass proteomic degradation and therefore increases the CFTR proteins trafficking to the cell surface in patients with this mutation.28
The combination therapy of ivacaftor and lumacaftor has produced considerable improvements in lung function, weight gain, and reduced exacerbations of pulmonary symptoms.53 The weight increase observed under this combination is speculated to be due to both improvement in pulmonary function and improvements in GI physiology.53
It is speculated that effective treatment of CFLD consists of adjuvant approach that repairs the primary defect and decreases inflammation.1 Nuclear receptors that modulate inflammation might represent a novel strategy to control inflammation seen in the CF biliary epithelium.1
Several other new CF medications are in clinical trials.12 Ataluren (PTC124), an orally bioavailable small molecule, designed to induce ribosomes to selectively read through premature stop codons during mRNA translation to produce functional CFTR.55 As nonsense mutations account for 10% of CFTR mutations, this modality can be an example of personalized medicine, with which patients with specific gene mutations are targeted with a specific modality of therapy.23
Conclusion
CFLD is common and considered to be the third leading cause of mortality in patients with CF after respiratory and transplantation complications. The clinical picture of CFLD can take many forms, ranging from mild asymptomatic hypertransaminasemia to cirrhosis and end-stage liver disease. Once the diagnosis of CFLD is made, annual follow-up by gastroenterologists or hepatologists is recommended. There is no effective therapy to prevent or treat CFLD. Management of CFLD emphasizes on optimizing nutritional status, avoiding hepatotoxic medications, and UDCA. Indications for liver transplant in CFLD include decompensated cirrhosis, with acceptable long-term outcomes. Novel therapy should consist of an adjuvant approach that repairs the primary defect and decreases inflammation. Effect of some of newer novel therapy approaches on CFLD is under clinical trials.
Disclosure
The authors declared no conflicts of interest. No funding was received for this study.
references
- 1. Fiorotto R, Strazzabosco M. Cystic fibrosis–related liver diseases: new paradigm for treatment based on pathophysiology. Clin Liver Dis (Hoboken) 2016 Nov;8(5):113-116.
- 2. Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005 May;352(19):1992-2001.
- 3. Wilschanski M, Novak I. The cystic fibrosis of exocrine pancreas. Cold Spring Harb Perspect Med 2013 May;3(5):a009746.
- 4. Kelly T, Buxbaum J. Gastrointestinal manifestations of cystic fibrosis. Dig Dis Sci 2015 Jul;60(7):1903-1913.
- 5. De Lisle RC, Borowitz D. The cystic fibrosis intestine. Cold Spring Harb Perspect Med 2013 Sep;3(9):a009753.
- 6. Castellani C, Duff AJ, Bell SC, Heijerman HG, Munck A, Ratjen F, et al. ECFS best practice guidelines: the 2018 revision. J Cyst Fibros 2018 Mar;17(2):153-178.
- 7. Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947-2003. Eur Respir J 2007 Mar;29(3):522-526.
- 8. Debray D, Kelly D, Houwen R, Strandvik B, Colombo C. Best practice guidance for the diagnosis and management of cystic fibrosis-associated liver disease. J Cyst Fibros 2011 Jun;10(Suppl 2):S29-S36.
- 9. Vo HD, Xu J, Rabinowitz SS, Fisher SE, Schwarz SM. The liver in pediatric gastrointestinal disease. J Pediatr Gastroenterol Nutr 2014 Sep;59(3):288-299.
- 10. Leeuwen L, Fitzgerald DA, Gaskin KJ. Liver disease in cystic fibrosis. Paediatr Respir Rev 2014 Mar;15(1):69-74.
- 11. Kamal N, Surana P, Koh C. Liver disease in patients with cystic fibrosis. Curr Opin Gastroenterol 2018 May;34(3):146-151.
- 12. Lindblad A, Glaumann H, Strandvik B. Natural history of liver disease in cystic fibrosis. Hepatology 1999 Nov;30(5):1151-1158.
- 13. Colombo C, Battezzati PM, Crosignani A, Morabito A, Costantini D, Padoan R, et al. Liver disease in cystic fibrosis: A prospective study on incidence, risk factors, and outcome. Hepatology 2002 Dec;36(6):1374-1382.
- 14. Colombo C. Hepatobiliary disease in cystic fibrosis. In: Kelly D, editor. Diseases of the liver and biliary system in children. 3rd ed. Oxford: Wiley-Blackwell; 2008. p. 270-288.
- 15. Colombo C, Russo MC, Zazzeron L, Romano G. Liver disease in cystic fibrosis. J Pediatr Gastroenterol Nutr 2006 Jul;43(Suppl 1):S49-S55.
- 16. Gooding I, Dondos V, Gyi KM, Hodson M, Westaby D. Variceal hemorrhage and cystic fibrosis: outcomes and implications for liver transplantation. Liver Transpl 2005 Dec;11(12):1522-1526.
- 17. Abdel-Salam E, Samuel S, Awad M, El-Marsafy A, Abdel-Meguid IE, Azmy J. Cystic fibrosis in Egyptian children: neonatal screening and high risk groups. JAC 1993;4(4):313-317.
- 18. Nazer HM. Early diagnosis of cystic fibrosis in Jordanian children. J Trop Pediatr 1992 Jun;38(3):113-115.
- 19. Al-Mahroos F. Cystic fibrosis in Bahrain incidence, phenotype, and outcome. J Trop Pediatr 1998 Feb;44(1):35-39.
- 20. Frossard PM, Girodon E, Dawson KP, Ghanem N, Plassa F, Lestringant GG, et al. Identification of cystic fibrosis mutations in the United Arab Emirates. Mutations in brief no. 133. Online. Hum Mutat 1998;11(5):412-413.
- 21. Nazer H, Rahbeeni Z. Cystic fibrosis and the liver–a Saudi experience. Ann Trop Paediatr 1994;14(3):189-194.
- 22. Alwan A, Modell B. Community control of genetic and congenital disorders. Alexandria: Eastern Mediterranean Regional Office, World Health Organization, Regional Office for the Eastern Mediterranean; 1997.
- 23. Ooi CY, Durie PR. Cystic fibrosis from the gastroenterologist’s perspective. Nat Rev Gastroenterol Hepatol 2016 Mar;13(3):175-185.
- 24. Diwakar V, Pearson L, Beath S. Liver disease in children with cystic fibrosis. Paediatr Respir Rev 2001 Dec;2(4):340-349.
- 25. Herrmann U, Dockter G, Lammert F. Cystic fibrosis-associated liver disease. Best Pract Res Clin Gastroenterol 2010 Oct;24(5):585-592.
- 26. Fustik S, Trajkovska M, Jakovska T, Spirevska L, Josifovska T, Koceva S. Screening for liver disease in cystic fibrosis: analysis of clinical and genetic risk factors for its development. Turk J Pediatr 2008 Nov-Dec;50(6):526-532.
- 27. Kobelska-Dubiel N, Klincewicz B, Cichy W. Liver disease in cystic fibrosis. Prz Gastroenterol 2014;9(3):136-141.
- 28. Kuk K, Taylor-Cousar JL. Lumacaftor and ivacaftor in the management of patients with cystic fibrosis: current evidence and future prospects. Ther Adv Respir Dis 2015 Dec;9(6):313-326.
- 29. Fiorotto R, Scirpo R, Trauner M, Fabris L, Hoque R, Spirli C, et al. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4-NF-κB-mediated inflammatory response in mice. Gastroenterology 2011 Oct;141(4):1498-1508, 1508.e1-1508.e5.
- 30. Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell 2006 Oct;11(4):441-450.
- 31. Gilad S, Meiri E, Yogev Y, Benjamin S, Lebanony D, Yerushalmi N, et al. Serum microRNAs are promising novel biomarkers. PLoS One 2008 Sep;3(9):e3148.
- 32. Szabo G, Bala S. MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol 2013 Sep;10(9):542-552.
- 33. Bala S, Petrasek J, Mundkur S, Catalano D, Levin I, Ward J, et al. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology 2012 Nov;56(5):1946-1957.
- 34. Cook NL, Pereira TN, Lewindon PJ, Shepherd RW, Ramm GA. Circulating microRNAs as noninvasive diagnostic biomarkers of liver disease in children with cystic fibrosis. J Pediatr Gastroenterol Nutr 2015 Feb;60(2):247-254.
- 35. Calvopina DA, Coleman MA, Lewindon PJ, Ramm GA. Function and regulation of micrornas and their potential as biomarkers in paediatric liver disease. Int J Mol Sci 2016 Oct;17(11):E1795.
- 36. Patriquin H, Lenaerts C, Smith L, Perreault G, Grignon A, Filiatrault D, et al. Liver disease in children with cystic fibrosis: US-biochemical comparison in 195 patients. Radiology 1999 Apr;211(1):229-232.
- 37. Lenaerts C, Lapierre C, Patriquin H, Bureau N, Lepage G, Harel F, et al. Surveillance for cystic fibrosis-associated hepatobiliary disease: early ultrasound changes and predisposing factors. J Pediatr 2003 Sep;143(3):343-350.
- 38. Durieu I, Pellet O, Simonot L, Durupt S, Bellon G, Durand DV, et al. Sclerosing cholangitis in adults with cystic fibrosis: a magnetic resonance cholangiographic prospective study. J Hepatol 1999 Jun;30(6):1052-1056.
- 39. Aqul A, Jonas MM, Harney S, Raza R, Sawicki GS, Mitchell PD, et al. Correlation of transient elastography with severity of cystic fibrosis-related liver disease. J Pediatr Gastroenterol Nutr 2017 Apr;64(4):505-511.
- 40. Klotter V, Gunchick C, Siemers E, Rath T, Hudel H, Naehrlich L, et al. Assessment of pathologic increase in liver stiffness enables earlier diagnosis of CFLD: results from a prospective longitudinal cohort study. PLoS One 2017 Jun;12(6):e0178784.
- 41. Koh C, Sakiani S, Surana P, Zhao X, Eccleston J, Kleiner DE, et al. Adult-onset cystic fibrosis liver disease: diagnosis and characterization of an underappreciated entity. Hepatology 2017 Aug;66(2):591-601.
- 42. Palaniappan SK, Than NN, Thein AW, Moe S, van Mourik I. Interventions for preventing and managing advanced liver disease in cystic fibrosis. Cochrane Database Syst Rev 2017 Aug;8:CD012056.
- 43. Smith JL, Lewindon PJ, Hoskins AC, Pereira TN, Setchell KD, O’Connell NC, et al. Endogenous ursodeoxycholic acid and cholic acid in liver disease due to cystic fibrosis. Hepatology 2004 Jun;39(6):1673-1682.
- 44. Cheng K, Ashby D, Smyth RL. Ursodeoxycholic acid for cystic fibrosis-related liver disease. Cochrane Database Syst Rev 2017 Sep;9:CD000222.
- 45. Siano M, De Gregorio F, Boggia B, Sepe A, Ferri P, Buonpensiero P, et al. Ursodeoxycholic acid treatment in patients with cystic fibrosis at risk for liver disease. Dig Liver Dis 2010 Jun;42(6):428-431.
- 46. McKeon D, Day A, Parmar J, Alexander G, Bilton D. Hepatocellular carcinoma in association with cirrhosis in a patient with cystic fibrosis. J Cyst Fibros 2004 Aug;3(3):193-195.
- 47. Kelleher T, Staunton M, O’Mahony S, McCormick PA. Advanced hepatocellular carcinoma associated with cystic fibrosis. Eur J Gastroenterol Hepatol 2005 Oct;17(10):1123-1124.
- 48. Melzi ML, Kelly DA, Colombo C, Jara P, Manzanares J, Colledan M, et al; EGSLTCF; European Liver Transplant Association (ELTA); European Cystic Fibrosis Society (ECFS). Liver transplant in cystic fibrosis: a poll among European centers. A study from the European Liver Transplant Registry. Transpl Int 2006 Sep;19(9):726-731.
- 49. Yi SG, Burroughs SG, Loebe M, Scheinin S, Seethamraju H, Jyothula S, et al. Combined lung and liver transplantation: analysis of a single-center experience. Liver Transpl 2014 Jan;20(1):46-53.
- 50. Harring TR, Nguyen NT, Liu H, Karpen SJ, Goss JA, O’Mahony CA. Liver transplantation in cystic fibrosis: a report from Baylor College of Medicine and the Texas Children’s Hospital. Pediatr Transplant 2013 May;17(3):271-277.
- 51. Mendizabal M, Reddy KR, Cassuto J, Olthoff KM, Faust TW, Makar GA, et al. Liver transplantation in patients with cystic fibrosis: analysis of United Network for Organ Sharing data. Liver Transpl 2011 Mar;17(3):243-250.
- 52. Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al; VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011 Nov;365(18):1663-1672.
- 53. Wainwright CE, Elborn JS, Ramsey BW. Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med 2015 Oct;373(18):1783-1784.
- 54. Harrison MJ, Murphy DM, Plant BJ. Ivacaftor in a G551D homozygote with cystic fibrosis. N Engl J Med 2013 Sep;369(13):1280-1282.
- 55. Ooi CY, Nightingale S, Durie PR, Freedman SD. Ursodeoxycholic acid in cystic fibrosis-associated liver disease. J Cyst Fibros 2012 Jan;11(1):72-73, author reply 74-75.