Pallister-Killian mosaic syndrome (PKS), also termed Pallister-Killian syndrome, Pallister mosaic aneuploidy, tetrasomy 12p mosaicism, isochromosome 12p syndrome, or Teschler-Nicola syndrome is a rare dysmorphic disorder caused by mosaic tetrasomy of the short arm of chromosome 12 (12p). It was first described by Pallister et al,1 and Teschler-Nicola and Killian.2
It is a multisystem disorder characterized by hypotonia in infancy and early childhood, intellectual disability, distinctive facial features, sparse hair (particularly around the temples), pigmentation, and other congenital disabilities. The disorder may be underdiagnosed, especially in those with mild features. More than 200 cases have been reported in the literature.
The presence of isochromosome 12p (made up of two p arms) causes the disorder. In patients with the syndrome, some cells have the two normal copies of chromosome 12, but other cells have the isochromosome 12p. These cells have four copies of all the genes on the p arm of chromosome 12. The extra genetic material of the isochromosome disrupts the normal development and causes the characteristic clinical features of the disorder.3–5
Case report
We report a case of an Omani female patient who was diagnosed with PKS at the National Genetics Center, Royal Hospital, Muscat, Oman. She was born preterm at 35 weeks gestation to non-consanguineous Omani parents. She was noted antenatally to have skeletal dysplasia and bowel herniation. She was born with multiple congenital anomalies including anterior abdominal wall defect, bowel herniation, and vestibular anus.6,7 She had craniofacial manifestations including a ‘coarse’ face with a flat profile, high forehead with temporal-frontal balding, sparse eyebrows and lashes, shallow supraorbital ridges, upslanting palpebral fissures, hypertelorism, and upturned nares. Her skeletal deformities were described as distal arthrogryposis of all limbs, talipes equinovalgus, and rhizomelic upper limbs, which were confirmed on plain X-rays.8
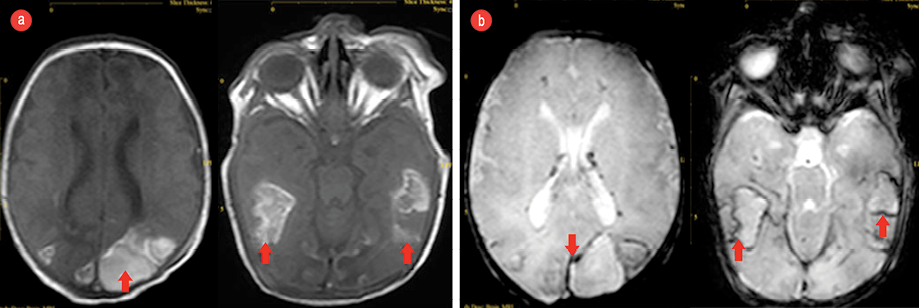
Figure 1: (a) Brain magnetic resonance imaging (MRI, T1-weighted) showing subacute hemorrhage (red arrows) in the temporal areas on both sides and (b) hemosiderin deposition around the hemorrhage (red arrows) seen via gradient SENSE MRI .
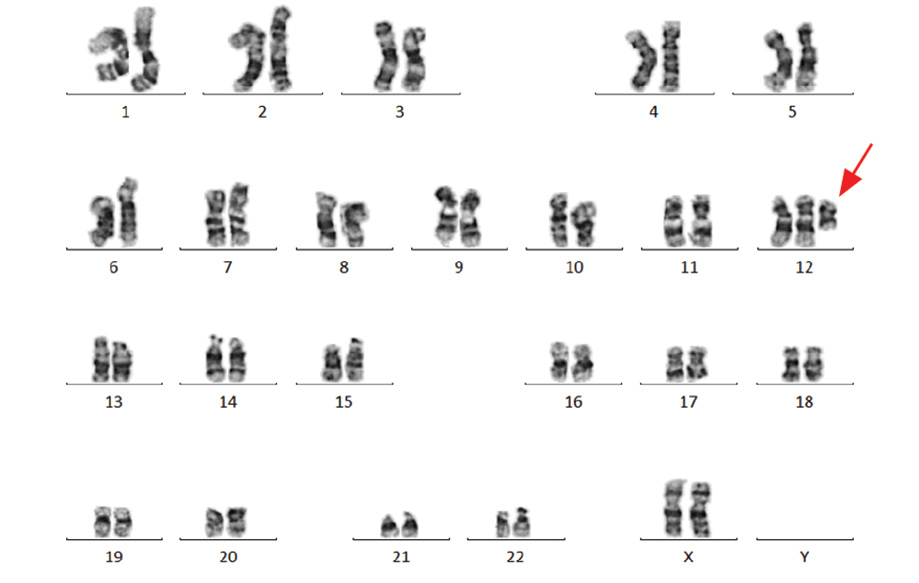
Figure 2: Karyotype image of tetrasomy 12p, obtained from peripheral lymphocytes showing the extra 12p isochromosome, indicated by the red arrow.
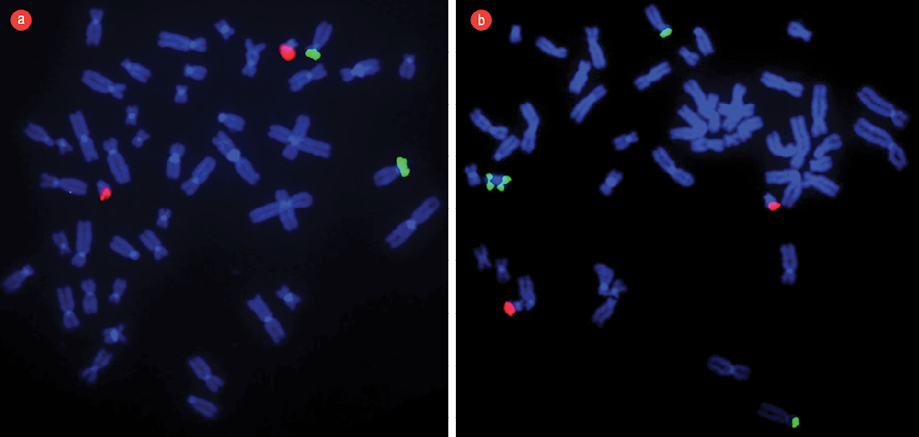
Figure 3: (a) Fluorescent in situ hybridization (FISH) technique applied in a normal pattern of chromosomes obtained from peripheral lymphocytes of a blood sample from a normal/healthy child. The dual color translocation probes [TEL/AML1] were used to show two green signals (TEL) at chromosome 12p and two orange signals (AML1) at chromosome 21p22 as contrast. (b) FISH technique applied to a pattern of chromosomes obtained from peripheral lymphocytes of a blood sample from our patient with Pallister-Killian syndrome. There were four green signals and two orange signals indicating the presence of 4-arms of chromosome 12p [green] and two signals for chromosome 21p22 [orange]. FISH technique confirmed the presence of iso(12p) and described in the karyotype as 47, XX,+i(12)(p10)[4]/46,XX[18].
She had a full screening for internal structural abnormalities. Her abdominal ultrasound was reported normal with the exception of mild echogenic kidneys. Echocardiography revealed cardiomegaly and abnormal heart configuration, 2.5 mm patent ductus arteriosus, with a left to right shunt and interrupted inferior vena cava. Brain computed tomography showed diffuse posterior brain and temporal lobes white matter edema. The posterior horns of the lateral brain ventricles were disproportionately dilated. A non-contrast magnetic resonance imaging showed late subacute intraparenchymal and intraventricular hemorrhages [Figure 1].
She had feeding difficulties, and was hypotonic with multiple contractures. This triggered neurology and metabolic investigations, which were mostly negative. Three days after delivery, she was subjected to surgery during which it was observed that gut rotation did not take place and the cecum and appendix were at the midline. The abdominal wall defect was closed, and rectal dilatation was performed.
The clinical features of this patient were severe; she had multiple congenital disabilities not compatible with life. She remained hospitalized for three months and was readmitted with severe aspiration pneumonia at four months of age. She remained hypotonic and was fed by a nasogastric tube and had severe global developmental delays. She died suddenly at the age of six months.
Blood samples in lithium heparin were cultured for 72 hours in RPMI-1640 medium (Gibco-BRL, USA) containing 20% fetal bovine serum (Gibco-BRL, USA) and antibiotics. The trypsin-Giemsa banding technique was used to analyze and categorize the chromosomes of 20 metaphase cells according to the International System for Human Cytogenetic Nomenclature (ISCN 2016).9
Chromosomal analysis of 72-hour peripheral blood lymphocytes cultures revealed a mosaic cell line. An abnormal clone (20% metaphase cells) showed the presence of an anomalous extra isochromosome 12p, the short arm of chromosome 12, indicating tetrasomy 12p. The remaining 80% of metaphase cells showed a normal 46, XX karyotype [Figure 2]. Fluorescent in situ hybridization (FISH) was applied to confirm the presence of duplication of chromosome 12p using the LSI TEL/AML1 ES dual color translocation probe (Abbott Laboratories, Il, USA). The presence of i(12) (p10) and tetrasomy 12p was confirmed by the detection of four green signals for 12p in 20% of analyzed cells [Figure 3]. The presence of iso(12p) has been expressed in the karyotype as: 47, XX,+i(12)(p10)[4]/46,XX[18].
Discussion
PKS is a sporadic, non-inherited cause of intellectual disability with a variable spectrum of congenital anomalies. It is a multisystem disorder characterized by hypotonia in infancy and early childhood, distinctive facial features, sparse hair (particularly around the temples), pigmentation and other multiple congenital disabilities including congenital heart defects, congenital diaphragmatic hernia, intellectual disability, and epilepsy.10,11 The parental origin of genetic errors are mostly maternal.12 The incidence of the syndrome is estimated at five per million live births.4,13 More than 200 cases have been reported in the literature.14 No ethnic group is spared, and PKS has been reported in almost every continent.15–18 The clinical features are highly variable, ranging from mild to severe. Normally, fetuses with PKS are aborted. However, the combination of mosaicism19 and complete or partial duplication of chromosome 12, leads to diverse phenotypes. The clinical presentations vary from severe fatal congenital anomalies at birth, to mild intellectual disabilities in adults.
Structural brain abnormalities in patients with severe PKS have been described in imaging studies.8,20,21 The main features include cerebral volume loss, malformations of the cortex, dysgenesis of the corpus callosum, and craniofacial malformations. Macrencephaly and cortical dysplasia are the likely explanation for the intellectual disability and epilepsy seen in patients.
Poulton et al,21 reviewed the literature for cases of PKS in which an intracranial scan was performed, and their observation was that 77% (72/93) of cases had a structural brain abnormality. Of these, ventricular abnormalities (46%; 33/72), abnormalities of the corpus callosum (25%; 18/72), and cerebral atrophy (29%; 21/72) were the most frequently reported. In few genetic studies, an attempt has been made to define minimal critical regions on chromosome 12p and their constituent genes associated with PKS.20,22
The clinical diversity and degree of severity of the disorder may lead to missed diagnosis of PKS, especially in those with mild features. Diagnosis can also be missed because of the low level of mosaicism in peripheral lymphocytes. The percentage of lymphocytes with the duplicate chromosome may be so low and it can be missed. Therefore, it is necessary to examine fibroblasts from the skin or buccal mucosa to confirm the diagnosis.23,24 These two issues have some serious implications for both the diagnosis and counseling of patients and families.8,13
The laboratory diagnosis of PKS in this patient depended on detection of the extra numerary 12p using classical peripheral lymphocyte karyotyping. This was quite surprising since mosaicism in PKS is rarely diagnosed in peripheral lymphocytes.3 Most cases of PKS in the literature are diagnosed using amniotic cells or skin fibroblasts. However, the clinical picture in this patient is similar to the severe forms of PKS in the literature. The craniofacial features and skeletal deformities were no different from those described previously. However, this infant was born with some other congenital anomalies of gut rotation including anterior abdominal wall defect, bowel herniation, and vestibular anus described only in the severest forms of PKS.6,7 Echocardiography showed cardiomegaly and abnormal heart configuration, patent ductus arteriosus and interrupted inferior vena cava.
Conclusion
We reported a case of PKS diagnosed in an Omani newborn patient. The clinical features in the reported patient were severe with multiple congenital disabilities. They were confirmed clinically, radiologically and during surgery and were not compatible with life. FISH study confirmed the presence of chromosome 12p duplication with the karyotype 47, XX,+i(12)(p10)[4]/46,XX[18]. This is the first case reported in the literature from Oman and the Arab world.
Disclosure
The authors declared no conflicts of interest.
references
- 1. Pallister PD, Meisner LF, Elejalde BR, Francke U, Herrmann J, Spranger J, et al. The pallister mosaic syndrome. Birth Defects Orig Artic Ser 1977;13(3B):103-110.
- 2. Teschler-Nicola M, Killian W. Case report 72: mental retardation, unusual facial appearance, abnormal hair. Synd Ident 1981;7:6-7.
- 3. Izumi K, Krantz ID. Pallister-Killian syndrome. Am J Med Genet C Semin Med Genet 2014 Dec;166C(4):406-413.
- 4. Blyth M, Maloney V, Beal S, Collinson M, Huang S, Crolla J, et al. Pallister-Killian syndrome: a study of 22 British patients. J Med Genet 2015 Jul;52(7):454-464.
- 5. Costa LS, Zandona-Teixeira AC, Montenegro MM, Dias AT, Dutra RL, Honjo RS, et al. Cytogenomic delineation and clinical follow-up of 10 Brazilian patients with Pallister-Killian syndrome. Mol Cytogenet 2015 Jun;8:43.
- 6. Lin AE, Clemens M, Garver KL, Wenger SL, Steele MW. Case of Pallister-Killian syndrome with imperforate anus. Am J Med Genet 1988 Nov;31(3):705-707.
- 7. Baglaj M, King J, Carachi R. Pallister-Killian syndrome: a report of 2 cases and review of its surgical aspects. J Pediatr Surg 2008 Jun;43(6):1218-1221.
- 8. Wilkens A, Liu H, Park K, Campbell LB, Jackson M, Kostanecka A, et al. Novel clinical manifestations in Pallister-Killian syndrome: comprehensive evaluation of 59 affected individuals and review of previously reported cases. Am J Med Genet A 2012 Dec;158A(12):3002-3017.
- 9. McGowan-Jordan J, Simons A, Schmid M, editors. ISCN 2016: An international system for human cytogenomic nomenclature. Publisher: Karger AG, Basel; 2016.
- 10. Cerminara C, Compagnone E, Bagnolo V, Galasso C, Lo-Castro A, Brinciotti M, et al. Late-onset epileptic spasms in children with Pallister-Killian syndrome: a report of two new cases and review of the electroclinical aspects. J Child Neurol 2010 Feb;25(2):238-245.
- 11. Candee MS, Carey JC, Krantz ID, Filloux FM. Seizure characteristics in Pallister-Killian syndrome. Am J Med Genet A 2012 Dec;158A(12):3026-3032.
- 12. Struthers JL, Cuthbert CD, Khalifa MM. Parental origin of the isochromosome 12p in Pallister-Killian syndrome: molecular analysis of one patient and review of the reported cases. Am J Med Genet 1999 May;84(2):111-115.
- 13. Roberts W, Zurada A, Zurada-ZieliŃSka A, Gielecki J, Loukas M. Anatomy of trisomy 12. Clin Anat 2016 Jul;29(5):633-637.
- 14. Pallister-Killian mosaic syndrome. US National Library of Medicine. Genetics Home Reference. [cited 2018 April]. Available from: https://ghr.nlm.nih.gov/condition/pallister-killian-mosaic-syndrome.
- 15. Desseauve D, Legendre M, Dugué-Maréchaud M, Vequeau-Goua V, Pierre F. [Prenatal and clinicopathological study of 6 cases of Pallister-Killian syndrome and review]. Gynecol Obstet Fertil 2016 Apr;44(4):200-206.
- 16. Toledo-Bravo de Laguna L, del Campo-Casanelles M, Santana-Rodriguez A, Santana-Artiles A, Sebastian-Garcia I, Cabrera-Lopez JC. [Three cases of Pallister-Killian syndrome]. Rev Neurol 2014 Jan;58(2):63-68.
- 17. Hasegawa T. [Pallister-Killian syndrome]. Ryoikibetsu Shokogun Shirizu 2000;(30 Pt 5):26-29.
- 18. Woodman BF, Jordan MA, Moller LI, Cartwright JD, De Ravel TJ. The Pallister-Killian syndrome in an African individual. Genet Couns 1995;6(1):33-36.
- 19. Spinner NB, Conlin LK. Mosaicism and clinical genetics. Am J Med Genet C Semin Med Genet 2014 Dec;166C(4):397-405.
- 20. Barkovich EJ, Lateef TM, Whitehead MT. Neuroimaging findings in Pallister-Killian syndrome. Neuroradiol J 2018 Aug;31(4):403-411.
- 21. Poulton C, Baynam G, Yates C, Alinejad-Rokny H, Williams S, Wright H, et al. A review of structural brain abnormalities in Pallister-Killian syndrome. Mol Genet Genomic Med 2018 Jan;6(1):92-98.
- 22. Izumi K, Conlin LK, Berrodin D, Fincher C, Wilkens A, Haldeman-Englert C, et al. Duplication 12p and Pallister-Killian syndrome: a case report and review of the literature toward defining a Pallister-Killian syndrome minimal critical region. Am J Med Genet A 2012 Dec;158A(12):3033-3045.
- 23. Manasse BF, Lekgate N, Pfaffenzeller WM, de Ravel TJ. The Pallister-Killian syndrome is reliably diagnosed by FISH on buccal mucosa. Clin Dysmorphol 2000 Jul;9(3):163-165.
- 24. Genevieve D, Cormier-Daire V, Sanlaville D, Faivre L, Gosset P, Allart L, et al. Mild phenotype in a 15-year-old boy with Pallister-Killian syndrome. Am J Med Genet A 2003 Jan;116A(1):90-93.