Kikuchi disease, or Kikuchi Fujimoto disease (KFD), is an uncommon benign illness first described by Japanese pathologists in 1972.1,2 The exact etiology is unknown, but viral and autoimmune causes have been suggested. It predominantly affects young adults of Asian ethnicity although cases have been reported worldwide.3 The typical clinical presentation includes fever and cervical lymphadenopathy, with the skin being the most affected extranodal organ, although such cases are rare. The clinical and laboratory findings of KFD can resemble systemic lupus erythematosus (SLE). As the disease is self-limiting, there is no specific treatment. Supportive therapy is typically employed, and corticosteroids may be initiated in severe cases.4
Case Report

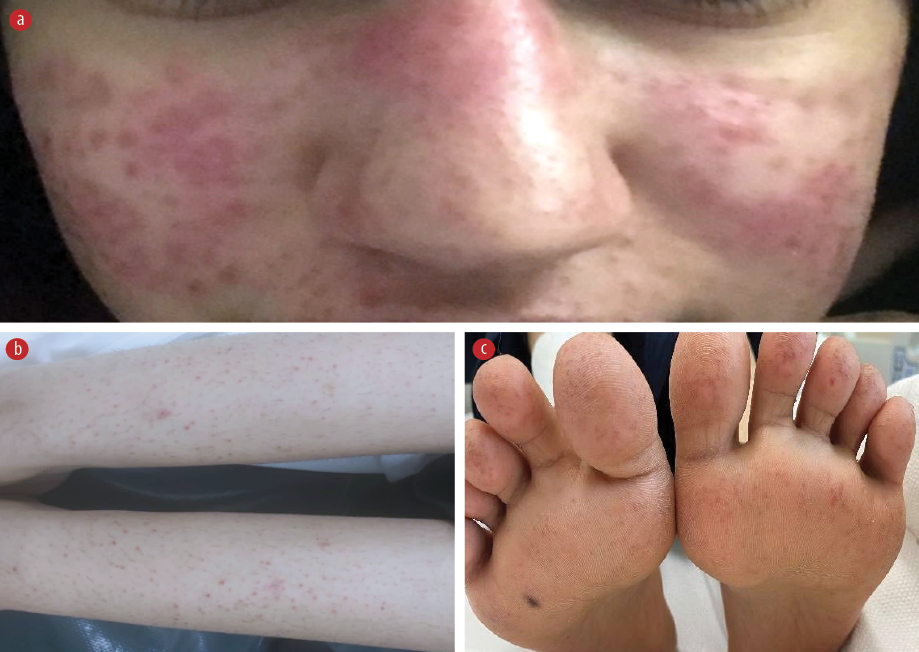 Figure 1: (a) Dusky to erythematous papules coalescing into a plaque over the malar area and dorsal nose. Petechiae and purpura on both forearms and (b) shins with involvement of the palm and (c) soles.
Figure 1: (a) Dusky to erythematous papules coalescing into a plaque over the malar area and dorsal nose. Petechiae and purpura on both forearms and (b) shins with involvement of the palm and (c) soles.

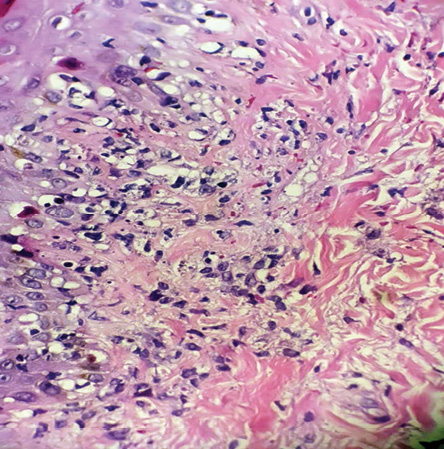 Figure 2: Skin biopsy show papillary and reticular lymphohistiocytic infiltration, interface dermatitis, and karyorrhexis (hematoxylin and eosin staining, magnification = 40 ×).
Figure 2: Skin biopsy show papillary and reticular lymphohistiocytic infiltration, interface dermatitis, and karyorrhexis (hematoxylin and eosin staining, magnification = 40 ×).
A 17-year-old female with no significant medical history presented to the emergency department with a high-grade fever, skin eruption lasting nearly one month, associated with weight loss of approximately 3 kg, malaise, arthralgia, and progressive swelling on the left side of her neck. Despite multiple courses of broad-spectrum antibiotics, there was no clinical improvement. The patient was admitted for further evaluation of her febrile illness.
On examination, the patient appeared sick, with fever and tachycardic. Physical examination revealed multiple palpable cervical lymph nodes on the left side, tender to touch. The largest swelling measured approximately 2 × 3 cm at level 3. There was an erythematous, non-blanching, non-scaly maculopapular eruption on the face, particularly affecting the malar area [Figure 1a]. Multiple non-pruritic, non-blanchable pinpoint erythematous macular eruptions were also observed on the forearms and lower extremities [Figure 1b] extending to the palms and soles [Figure 1c]. No hepatosplenomegaly or neurological deficits were detected. The rest of her systemic examination was unremarkable.
Laboratory studies showed the following results: hemoglobin was 8.5 g/dL, white blood cell count was 1.99 × 109/L, platelets count was 568 × 109, the international normalized ratio was 1.12 , prothrombin time was 12.8 s, activated partial thromboplastin time normal range, Ferritin was 571 ug/L, lactate dehydrogenase was 515 U/L, C-reactive protein was 37 mg/L, alanine transaminase was 66 U/L, and aspartate aminotransferase was 107 U/L. Hepatitis B surface antigen and hepatitis C antibodies were negative, C3 was 2.24 g/L, and C4 was 0.41 g/L. Antinuclear antibodies were detected with a titer of 1/640 and a speckled pattern. Anti-double stranded DNA and extractable nuclear antigen were negative. Blood, urine analysis, and cultures were also negative. Throat swab, COVID-19 rapid antigen test, and Quantiferon test (Interferon-Gamma Release Assay) were all negative. HIV and syphilis test were negative. Epstein–Barr virus serology showed positive immunoglobulin G and Epstein–Barr virus nuclear antigen, but negative for immunoglobulin M. Other viral and bacterial serological tests yielded negative results.
Imaging studies were done, including an echocardiogram and chest X-ray, which were both normal. A pan computed tomography scan was performed due to suspicion of lymphoproliferative disease. The scan showed multiple enhancing lymph nodes in the left submandibular, left upper, mid, deep cervical, and posterior cervical regions. Some of these lymph nodes showed central necrosis. Bilateral axillary lymph nodes were small and non-necrotic. No lymphadenopathy was found in the abdomen and pelvis.
To rule out other potential causes, excisional lymph node biopsy and skin biopsy were performed. The lymph node biopsy showed extensive necrosis composed of histiocytes, plasmacytoid dendritic cells, and abundant karyorrhectic debris. No neutrophils or plasma cells were seen. No atypical lymphoid infiltration or granuloma were seen. Immunohistochemistry revealed that the mononuclear cell infiltrate consisted of predominantly CD68-positive histocytes and CD3-positive T cells. The histocytes exhibited myeloperoxidase positivity, and the T-cells were mostly CD8-positive with a smaller number of CD4-positive cells. Epstein-Barr encoding region in situ hybridization showed negative staining for Epstein-Barr virus.
A skin biopsy was performed, revealing vacuolar interface changes and scattered necrotic keratinocytes. In the papillary and reticular dermis, there was perivascular and interstitial lymphohistiocytic infiltration with prominent karyorrhexis. No neutrophils, eosinophils, or plasma cells were seen [Figure 2]. Ziehl-Neelsen staining was negative for acid-fast bacilli. Immunohistochemistry showed perivascular and interstitial infiltration of CD68-positive histiocytes as the predominant inflammatory cells [Figure 3a] along with a higher number of CD8-positive cytotoxic T-lymphocytes were predominant cells of the lymphocytes compared to few CD4 cells [Figure 3b].
Considering the patient’s age, gender, clinical presentation, and the histopathological findings of the skin and lymph nodes, a diagnosis of KFD with skin involvement was made. The patient was started on oral prednisolone at 1 mg/kg. After one to three weeks of treatment, the fever resolved, lymphadenopathy improved to a non-palpable state, and the skin rash improved.

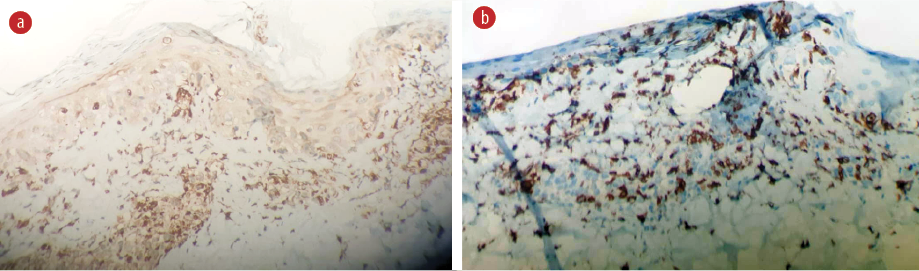 Figure 3: Immunohistochemistry (a) CD68 shows that histocytes are the predominant inflammatory cells. (b) CD-8 positive cytotoxic T-lymphocytes are the predominant cells of the lymphocytes (hematoxylin and eosin staining, magnification = 40 ×).
Figure 3: Immunohistochemistry (a) CD68 shows that histocytes are the predominant inflammatory cells. (b) CD-8 positive cytotoxic T-lymphocytes are the predominant cells of the lymphocytes (hematoxylin and eosin staining, magnification = 40 ×).
Discussion
KFD is a rare condition with unclear etiology and pathophysiology. Two theories have been proposed regarding the cause of KFD: infectious and autoimmune. Various bacterial and viral agents have been identified as potential triggers. Additionally, an association between KFD and autoimmune diseases, particularly SLE, has been observed.3 The exact relationship between SLE and KFD is not fully understood.5 Both diseases predominantly affect young women, and they share several
clinical characteristics.6
Our patient had common symptoms and laboratory findings of SLE, such as fever, malaise, weight loss, arthralgia, malar rash, cervical lymphadenopathy, anemia, leukopenia, and positive antinuclear antibodies.
The patient presented with a high platelet count, which gradually reduced to the normal range after starting oral prednisolone. In a recent study looking at blood changes in 367 patients with KFD, 282 (77%) had complete blood count data; of these, anemia (22%) was the most common abnormality, followed by lymphopenia (17%), neutropenia (11%), atypical lymphocytes (9%), and thrombocytopenia (8%) but the increased cell count was relatively rare.7 A case was reported in Turkey for a female patient diagnosed with KFD who suddenly had deranged hemoglobin and raised D-dimer, activated partial thromboplastin time, prothrombin time, and international normalized ratio with decreased fibrinogen and developed disseminated intravascular coagulopathy.8
Several studies have reported cases of KFD associated with SLE. Some patients were diagnosed with SLE following the diagnosis of KFD, while others had a prior diagnosis of SLE. Kucukardali et al,9 reported 28 cases of SLE-associated KFD that satisfied the diagnostic criteria for SLE in a study encompassing 244 patients; of those, 18 cases had both KFD and SLE, while six cases had SLE diagnosed after KFD, and four cases had a prior diagnosis of SLE. Santana et al,10 reported 35 cases of SLE-associated KFD, in which 14 patients were diagnosed with SLE following KFD. Patra et al,11 reported a case of a woman who developed SLE after two years of KFD. In four cases, KFD was identified before the diagnosis of SLE according to Goldblatt et al.12 Therefore, a close follow-up is recommended to detect the evolution of SLE.
In Oman, where our patient was from, only two cases of Kikuchi disease have been reported, with skin manifestations observed in one case.13,14 In Saudi Arabia, 15 cases were reported as KFD in one report,15 with another reporting five cases.16 There have been 14 cases documented in Qatar, three of which had cutaneous symptoms.17 There is limited data on whether these reported cases of KFD had a previous diagnosis of SLE or they developed SLE later.
Skin manifestations in KFD are non-specific and have been described in 40% of cases18 and may resemble lupus eruption. In our patient, skin eruption preceded the appearance of lymphadenopathy and improved after initiating treatment with oral prednisolone. Histopathological criteria, including the presence of karyorrhexis and the absence of neutrophils as major criteria, have been proposed to diagnose KFD with skin involvement. Minor criteria included the presence of interface dermatitis, the presence of inflammatory cell infiltration in the reticular dermis, or subcutaneous fat tissue.19 Our patient met all the major and minor criteria supporting the diagnosis of KFD.
Histopathological findings of cutaneous lesions in KFD can resemble those seen in SLE, such as interface dermatitis, dermal mucin deposition, and panniculitis.20 However, plasma cells, which are commonly observed in SLE, are usually absent in KFD.19
A definitive diagnosis of KFD can be made based on the histopathological findings from the lymph node biopsy, ruling out other possible causes. The histopathological features that can support SLE are an increased number of plasma cells, hematoxylin bodies, DNA deposits in the vascular walls, neutrophilic infiltration, and varying degrees of coagulative necrosis with the Azzopardi phenomenon. In contrast, the absence of hematoxylin bodies and neutrophils indicates KFD rather than SLE.20
As KFD is a self-limiting disease, no specific treatment is required, and the condition usually resolves within one to six months. Treatment is generally symptomatic treatment with analgesics and antipyretics. However, severe cases may benefit from a course of corticosteroids.4
Conclusion
Our case highlights the importance of considering KFD as a rare entity when evaluating patients with clinical features and laboratory findings that mimic SLE. The similarities between KFD and SLE in terms of clinical presentation and laboratory results make it challenging to differentiate between the two conditions without a biopsy. It is crucial to perform a histopathological examination, such as a lymph node biopsy, to confirm the diagnosis and exclude other potential causes. Proper diagnosis is essential to avoid inappropriate treatment and to ensure appropriate long-term follow-up, considering the possibility of the evolution of SLE.
Disclosure
The authors declared no conflicts of interest. Written consent was obtained from the patient.
references
- 1. Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytes: a clinicopathological study. Acta Haematol Jpn 1972;35:379-380.
- 2. Fujimoto Y. Cervical subacute necrotizing lymphadenitis. A new clinicopathological entity. Intern Med 1972;30:920-927.
- 3. Perry AM, Choi SM. Kikuchi-fujimoto disease: a review. Arch Pathol Lab Med 2018 Nov;142(11):1341-1346.
- 4. Masab M, Surmachevska N, Farooq H. Kikuchi disease. StatPearls Publishing 2022.
- 5. Londhey VA, Buche AS, Kini SH, Rajadhyaksha GC. Kikuchi fujimoto disease and systemic lupus erythematosus–a rare association. J Assoc Physicians India 2010 Oct;58:642-643.
- 6. Komócsi A, Tóvari E, Pajor L, Czirják L. Histiocytic necrotizing lymphadenitis preceding systemic lupus erythematosus. J Eur Acad Dermatol Venereol 2001 Sep;15(5):476-480.
- 7. Yu SC, Huang HH, Chen CN, Chen TC, Yang TL. Blood cell and marrow changes in patients with Kikuchi disease. Haematologica 2022 Aug;107(8):1981-1985.
- 8. Uslu E, Gurbuz S, Erden A, Aykas F, Karagoz H, Karahan S, et al. Disseminated intravascular coagulopathy caused by Kikuchi-fujimoto disease resulting in death: first case report in Turkey. Int Med Case Rep J 2014 Feb;7:19-22.
- 9. Kucukardali Y, Solmazgul E, Kunter E, Oncul O, Yildirim S, Kaplan M. Kikuchi-fujimoto disease: analysis of 244 cases. Clin Rheumatol 2007 Jan;26(1):50-54.
- 10. Santana A, Lessa B, Galrão L, Lima I, Santiago M. Kikuchi-Fujimoto’s disease associated with systemic lupus erythematosus: case report and review of the literature. Clin Rheumatol 2005 Feb;24(1):60-63.
- 11. Patra A, Bhattacharya SK. SLE developing in a follow-up patient of Kikuchi’s disease: a rare disorder. J Clin Diagn Res 2013 Apr;7(4):752-753.
- 12. Goldblatt F, Andrews J, Russell A, Isenberg D. Association of Kikuchi-fujimoto’s disease with SLE. Rheumatology (Oxford) 2008 Apr;47(4):553-554.
- 13. Ahmed S, Burney IA, Elnoor IB. Kikuchi’s disease in a 9-year-old girl: case report. Ann Trop Paediatr 2005 Sep;25(3):219-221.
- 14. Al Awfi MM, Al Badaai Y, Al Haddabi I. Unilateral cervical lymphadenopathy. Oman Med J 2020 Apr;35(2):e121.
- 15. Al-Maghrabi J, Kanaan H. Histiocytic necrotising lymphadenitis (Kikuchi-fujimoto disease) in Saudi Arabia: clinicopathology and immunohistochemistry. Ann Saudi Med 2005;25(4):319-323.
- 16. Kutty MK, Anim JT, Sowayan S. Histiocytic necrotising lymphadenitis (Kikuchi-fujimoto disease) in Saudi Arabia. Trop Geogr Med 1991;43(1-2):68-75.
- 17. Al Soub H, Al Bosom I, El Deeb Y, Al Maslamani M, Al KHuwaiter J. Histiocytic necrotizing lymphadenitis (Kikuchi-fujimoto disease): a study of 14 cases from Qatar. Qatar Medical Journal 2006 Nov;2006(2):13.
- 18. Kuo TT. Cutaneous manifestation of Kikuchi’s histiocytic necrotizing lymphadenitis. Am J Surg Pathol 1990 Sep;14(9):872-876.
- 19. Kim JH, Kim YB, In SI, Kim YC, Han JH. The cutaneous lesions of Kikuchi’s disease: a comprehensive analysis of 16 cases based on the clinicopathologic, immunohistochemical, and immunofluorescence studies with an emphasis on the differential diagnosis. Hum Pathol 2010 Sep;41(9):1245-1254.
- 20. Găman M, Vlădăreanu AM, Dobrea C, Onisâi M, Marinescu C, Voican I, et al. A Challenging case of Kikuchi-fujimoto disease associated with systemic lupus erythematosus and review of the literature. Case Rep Hematol 2018 Jan;2018:1791627.