With the turn of the twenty-first century and the use of modern genetic testing, the identification of dominant or recessive genetic mutations related to Parkinson’s disease (PD) is becoming more efficient. These tests uncover genetic mutations that play an important role in the early diagnosis of early-onset PD and juvenile PD. Between 3–5% of all patients with PD have onset before the age of 40 years,1 which is likely related to genetic causes.2 Parkin gene mutations are the most common and are associated with autosomal recessive early-onset PD.3 Patients with these types of mutations lose dopaminergic neurons in the substantia nigra and noradrenergic neurons in the locus coeruleus.4 We believe that we have discovered the first case of an autosomal recessive early-onset PD mutation related to the PARK2 gene with cysteine codon 201X (p. Cys201X) mutation.
Case report
The patient was a 34-year-old Emirati female who worked as a schoolteacher. She presented with complaints of limb and speech tremors. She explained her difficulties in initiating movement and speech during her teaching session. She had first noticed these difficulties two years ago with her right upper extremity being affected initially. With time, her symptoms progressed and become more evident. Her medical history was significant for focal to bilateral tonic-clonic seizures for the past three years, which was supported by an electroencephalography study demonstrating abnormal bitemporal epileptiform activity with secondary generalization. Her seizures were well controlled with oral levetiracetam 500 mg taken twice daily, and she has been free of seizures for more than one year. The patient has one sister and four brothers, all of whom are healthy. She is unaware of any family members with PD or any genetic disorders.
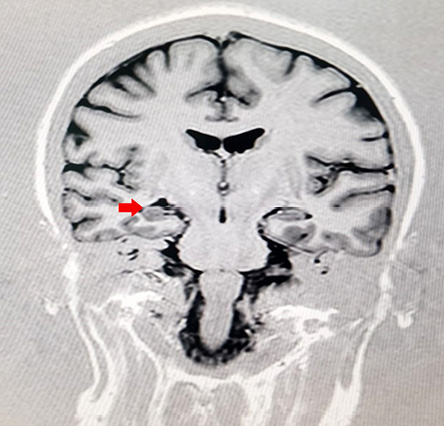
Figure 1: Magnetic resonance imaging epilepsy protocol of the patient showing right mesial temporal atrophy (red arrow).
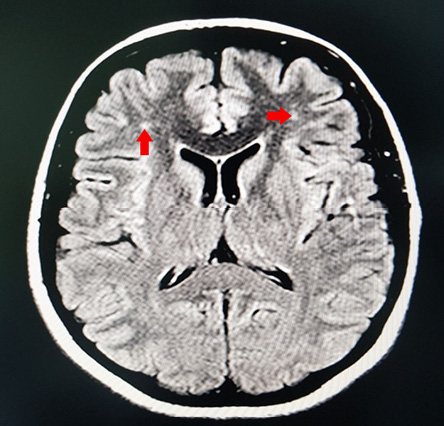
Figure 2: Magentic resonance imaging of the brain T2/FLAIR sequence showing nonspecific white spots in the frontal lobe (red arrows).
On examination, the patient seemed worrisome while sitting on the edge of the chair. She was oriented to time, place, and person. Her mini-mental score was 30/30, and her vital signs were stable. Her speech was noted to be tremulous and stuttering in character. Also, she had reduced eyelid blinking movements and hypomimia. Otherwise, cranial nerve examination was within normal limits. Her motor examination showed normal strength; however, she had severe bilateral upper and lower extremity rigidity, which was more evident on the right side. While resting, the patient exhibited bilateral pin-rolling tremors in both of her upper extremities. Her examination was notable for features of limb bradykinesia, including reduced leg tapping and slowly rising from the chair. In addition, her gait was shuffling in nature with reduced arm swinging and abnormal retropulsion.
A complete workup was done, including review of all her medications, brain imaging, vasculitis workup, in addition to an electrophysiological study, which were normal. All secondary causes of Parkinsonism disorders were ruled out. All of her laboratory results were normal, including total urine copper and ceruloplasmin levels. Magnetic resonance imaging and angiography of the brain revealed a right-sided mesial temporal atrophy [Figure 1] in addition to nonspecific white matter spots in the frontal lobes [Figure 2]. With the above findings in mind, a diagnosis of early-onset PD was suspected, and genetic testing was requested.
Genetic decoding was performed by Bioscientia - Center of Human Genetics in Ingelheim, Germany. Direct polymerase chain reaction sequencing of coding exons 1 to 12 was performed to analyze Parkin (PARK2) mutations. The results of the analysis indicated a 1 base pair (1-bp) insertion in exon 5 of PARK2 (c.601_602insA) predicting a nonsense mutation causing substitution of a cysteine codon by a stop codon (p. Cys201X) predicted to produce a truncated protein. Moreover, multiplex amplicon quantification showed the presence of this mutation on both exon 5 alleles, thus indicating homozygosity. It is likely that PARK2 has lost its function as a result of this premature stop codon. This mutation is not found in the Genome Aggregation Database (gnomAD).5 We believe this is a unique mutation as, to the best of our knowledge, it has never been described previously. The patient was started on levodopa 100 mg/carbidopa 25 mg and showed an excellent response to treatment within a year. On follow-up, one month later, the patient was noted to have an improvement in her mobility, tremor, and stiffness two-to-three hours after taking her medication. About six months later, she showed no rigidity or bradykinesia, and her tremor was much less than before. At one year follow-up, she was put on levodopa 100 mg/carbidopa 25 mg/entacapone 200 mg and pramipexol 1 mg thrice daily. At present, her symptoms are much improved compared to her initial presentation, and she is doing very well on treatment and regular follow-up.
Discussion
PD is one the most frequent neurodegenerative disorders characterized by bradykinesia, resting tremor, postural instability, and rigidity.6 Although the prevalence of this disorder is age-dependent, a number of recessive mutations have been identified to be associated with an early-onset,7 and, in some cases, a familial variant of this disorder.8,9 A systematic review has identified mutations in PRKN (PARK2/ Parkin), PINK1, and PARK7 (DJ1) accounting for nearly 13% of cases of early-onset PD.10 Out of these genes, mutations involving PARK2 gene are known to be the most common cause of autosomal recessive early-onset PD.10–12
In the human genome, the PARK2 gene is known to be one of the largest, with 12 exons spanning over 1.3 Mb encoding 465 amino acids.6 Mutations of PARK2 have been identified in more than half of the cases of autosomal recessive familial early-onset PD.3 Most cases of early-onset PD associated with PARK2 mutations have been identified to manifest phenotypically in a homozygous or a compound heterozygous state.3,6 This was also the case in our patient, where the mutation was found to be homozygous. There are inconclusive studies that show a single heterozygous mutation is insufficient to manifest phenotypically,12,13 although this may be a risk factor for late-onset PD.14
Although early-onset (< 45 years) and dystonia are reported to be common symptoms in autosomal recessive juvenile Parkinson’s;6 dystonia, dyskinesia, and motor fluctuations are reported to be more common in Parkin proven disease.10 Also, cognitive decline and atypical symptoms like pyramidal signs and spasticity are uncommon in patients with PARK2 mutations,3,10 as was also the case in our patient who had intact cognitive function. Loss of neurons in substantia nigra and locus coeruleus is most commonly reported in a neuropathological autopsy of such patients along with the absence of Lewy bodies, alpha-Synuclein, or Tau pathology.6,8 While impairment of olfaction is one of the earliest features of PD, olfaction is found to be commonly spared in patients with PARK2 mutation associated early-onset PD.15 Sleep disturbance is another feature that is frequently noticed among patients with PD and significantly affects their quality of life.16 Patients with early-onset PD may also experience similar symptoms as evidenced by a case of juvenile PD in the literature that was reported to have concurrent rapid eye movement sleep behavioral disorder as well.17
Usually, PARK2 mutation associated early-onset PD responds well to levodopa treatment, just like late-onset Parkinson’s.6,10 This was also the case with our patient, who showed excellent response to treatment. It must be noted that a significant proportion of patients do experience dyskinesia as a result of treatment with levodopa.10 This contrasts against patients with PARK7 (DJ1) mutation where the response to levodopa therapy is not as remarkable.18
Conclusion
We described a case of early-onset PD in a 34-year-old female who, on genetic analysis, was found to have a previously undescribed homozygous mutation in the PARK2 gene. The patient showed an excellent response to treatment. Given the slow and rather agonizing course of this disease and good response to treatment, screening for these mutations can be of significant benefit to patients and must be considered in patients and families with symptoms of PD appearing disproportionate to age.
Disclosure
The authors declared no conflicts of interest.
references
- 1. Schrag A, Schott JM. Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet Neurol 2006 Apr;5(4):355-363.
- 2. Poorkaj P, Nutt JG, James D, Gancher S, Bird TD, Steinbart E, et al. Parkin mutation analysis in clinic patients with early-onset Parkinson [corrected] disease. Am J Med Genet A 2004 Aug;129A(1):44-50.
- 3. Lücking CB, Dürr A, Bonifati V, Vaughan J, De Michele G, Gasser T, et al; French Parkinson’s Disease Genetics Study Group; European Consortium on Genetic Susceptibility in Parkinson’s Disease. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med 2000 May;342(21):1560-1567.
- 4. Ishikawa A, Takahashi H. Clinical and neuropathological aspects of autosomal recessive juvenile parkinsonism. J Neurol 1998 Nov;245(11)(Suppl 3):4-9.
- 5. gnomAD [Internet]. Gnomad.broadinstitute.org. 2020 [cited 2020 June 4]. Available from: https://gnomad.broadinstitute.org/gene/ENSG00000185345?dataset=exac.
- 6. Mata IF, Lockhart PJ, Farrer MJ. Parkin genetics: one model for Parkinson’s disease. Hum Mol Genet 2004 Apr;13(Spec No 1):R127-R133.
- 7. Puschmann A. Monogenic Parkinson’s disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord 2013 Apr;19(4):407-415.
- 8. Cornejo-Olivas MR, Torres L, Mata IF, Mazzetti P, Rivas D, Cosentino C, et al. A Peruvian family with a novel PARK2 mutation: Clinical and pathological characteristics. Parkinsonism Relat Disord 2015 May;21(5):444-448.
- 9. Shi Y, Kawakami H, Zang W, Li G, Zhang J, Xu C. Novel compound heterozygous mutations in the PARK2 gene identified in a Chinese pedigree with early-onset Parkinson’s disease. Brain Behav 2017 Dec;8(1):e00901.
- 10. Kasten M, Hartmann C, Hampf J, Schaake S, Westenberger A, Vollstedt EJ, et al. Genotype-phenotype relations for the parkinson’s disease genes parkin, pink1, dj1: mdsgene systematic review. Mov Disord 2018 May;33(5):730-741.
- 11. Periquet M, Latouche M, Lohmann E, Rawal N, De Michele G, Ricard S, et al; French Parkinson’s Disease Genetics Study Group; European Consortium on Genetic Susceptibility in Parkinson’s Disease. Parkin mutations are frequent in patients with isolated early-onset parkinsonism. Brain 2003 Jun;126(Pt 6):1271-1278.
- 12. Kay DM, Stevens CF, Hamza TH, Montimurro JS, Zabetian CP, Factor SA, et al. A comprehensive analysis of deletions, multiplications, and copy number variations in PARK2. Neurology 2010 Sep;75(13):1189-1194.
- 13. Lincoln SJ, Maraganore DM, Lesnick TG, Bounds R, de Andrade M, Bower JH, et al. Parkin variants in North American Parkinson’s disease: cases and controls. Mov Disord 2003 Nov;18(11):1306-1311.
- 14. Foroud T, Uniacke SK, Liu L, Pankratz N, Rudolph A, Halter C, et al; Parkinson Study Group. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology 2003 Mar;60(5):796-801.
- 15. Alcalay RN, Siderowf A, Ottman R, Caccappolo E, Mejia-Santana H, Tang MX, et al. Olfaction in Parkin heterozygotes and compound heterozygotes: the CORE-PD study. Neurology 2011 Jan;76(4):319-326.
- 16. Salawu F, Olokoba A. Excessive daytime sleepiness and unintended sleep episodes associated with Parkinson’s disease. Oman Med J 2015 Jan;30(1):3-10.
- 17. Rye DB, Johnston LH, Watts RL, Bliwise DL. Juvenile Parkinson’s disease with REM sleep behavior disorder, sleepiness, and daytime REM onset. Neurology 1999 Nov;53(8):1868-1870.
- 18. Taipa R, Pereira C, Reis I, Alonso I, Bastos-Lima A, Melo-Pires M, et al. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 2016 Jun;139(Pt 6):1680-1687.